
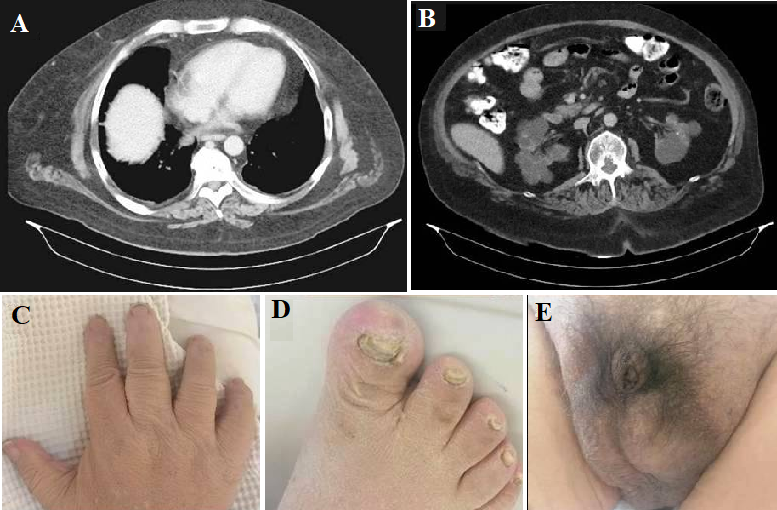
Mediastinal lipomatosis in a patient with Bardet - Biedl syndrome: more diverse than previously thought
Eleni Paschou, Nikolaos Sabanis
PAMJ. 2023; 45:82. Published 13 Jun 2023 | doi:10.11604/pamj.2023.45.82.35582
Corresponding author
Eleni Paschou, Department of General Practice and Family Medicine, Health Center of Aliartos, Levadia, Greece (el_paschou@yahoo.gr)
This image
| Articles published in JOURNAL_ABBREVIATION are Open Access and distributed under the terms of the Creative Commons Attribution 4.0 International (CC BY 4.0). |  |
eISSN: 1937-8688
The Pan African Medical Journal (ISSN: 1937-8688) is a subsidiary of the Pan African Medical Journal. The contents of this journal is intended exclusively for professionals in the medical, paramedical and public health and other health sectors.
Currently tracked by: DOAJ, AIM, Google Scholar, AJOL, EBSCO, Scopus, Embase, IC, HINARI, Global Health, PubMed Central, PubMed/Medline, ESCI
Physical address: "Kenya: 3rd Floor, Park Suite Building, Parkland Road, Nairobi. PoBox 38583-00100, tel: +254 (0)20-520-4356 | Cameroon: Immeuble TechnoPark Essos, Yaounde, PoBox: 10020 Yaounde, tel: +237 (0)24-309-5880"
PAMJ
Primary Health Care Practice Journal
African Journal of Health Economics, Systems and Policy
PAMJ Conference Management System

About PAMJ - Manuscript Hut™
The Manuscript Hut is a product of the PAMJ Center for Public health Research and Information.
Kenya: 3rd Floor, Park Suite Building, Parkland Road, Nairobi. PoBox 38583-00100, tel: +254 (0)20-520-4356
Cameroon: Immeuble TechnoPark Essos, Yaounde, PoBox: 10020 Yaounde, tel: +237 (0)24-309-5880
Copyright © - Pan African Medical Journal - CEPHRI (Kenya). 2026
PAMJ One Manuscript Hut - (MMS V.4.0). Release date April 2026 - Customized for PAMJ.
For advertisers Contact the PAMJ sales service. Download our latest media-kit.
sales-service@panafrican-med-journal.com
 |