
Alacrymie congénitale révélant un syndrome d’Allgrove: ŕ propos de trois cas
Rajae Derrar, Nourredinne Boutimzine, Amina Laghmari, Amal Alouane, Rajae Daoudi
Corresponding author: Rajae Derrar, Université Mohammed V Souissi, Service d’'Ophtalmologie A Hôpital des Spécialités CHU Rabat, Maroc 
Received: 30 May 2014 - Accepted: 22 Sep 2014 - Published: 14 Apr 2015
Domain: Clinical medicine
Keywords: Alacrymie congénitale, population pédiatrique, Achalasie śsophagienne
©Rajae Derrar et al. Pan African Medical Journal (ISSN: 1937-8688). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: Rajae Derrar et al. Alacrymie congénitale révélant un syndrome d’Allgrove: ŕ propos de trois cas. Pan African Medical Journal. 2015;20:359. [doi: 10.11604/pamj.2015.20.359.4717]
Available online at: https://www.panafrican-med-journal.com//content/article/20/359/full
Original article 
Alacrymie congénitale révélant un syndrome d’Allgrove: ŕ propos de trois cas
Alacrymie congénitale révélant un syndrome d’Allgrove: ŕ propos de trois cas
Rajae Derrar1,&, Nourredinne Boutimzine1, Amina Laghmari1, Amal Alouane1, Rajae Daoudi1
1Université Mohammed V Souissi, Service d’'Ophtalmologie A Hôpital des Spécialités CHU Rabat, Maroc
&Auteur correspondant
Rajae Derrar, Université Mohammed V Souissi, Service d’'Ophtalmologie A Hôpital des Spécialités CHU Rabat, Maroc
Le syndrome d'Allgrove ou triple A syndrome est une affection autosomique récessive constatée chez la population pédiatrique, associant dans sa forme complčte: Achalasie œsophagienne, Alacrymie, maladie d'Addison (insuffisance surrénale), une dégénérescence neurologique et occasionnellement une instabilité du systčme autonome. Nous rapportons les cas de 3 enfants issus de mariages consanguins, chez qui l'examen ophtalmologique a révélé une sécheresse sévčre avec dans deux cas une kératite envahissant l'axe visuel, ainsi qu'une paresse du reflexe photomoteur. Le bilan radiologique: transit œsogastroduodénal (TOGD) et fibroscopie œsogastroduodénale (FOGD) a révélé un mégaoesophage associé dans un cas ŕ une œsophagite. Un traitement ŕ base de larmes artificielles est instauré aussitôt, ainsi qu'un traitement chirurgical par voie laparoscopique. La connaissance de cette pathologie permettra une prise de conscience de la gravité de cette maladie en plus de suggérer sa prise en charge.
Le syndrome d'Allgrove ou syndrome des 3 A est une affection génétique de transmission autosomique récessive associant dans sa forme complčte : Achalasie œsophagienne, Alacrymie et insuffisance surrénale. C'est une affection rare, moins de 80 cas ont été rapportés dans la littérature [1].
Nous rapportons trois cas récents révélés par une alacrymie.
Observation 1
Garçon âgé de 4 ans, 2čme d'une fratrie de 5, issu d'un mariage consanguin consulte pour une baisse d'acuité visuelle bilatérale chiffrée ŕ 2/10čme au niveau de l'œil droit et ŕ 3/10čme faible au niveau de l'œil gauche non améliorable. L'examen ŕ la LAF trouve une kératopathie sčche (Figure 1) avec un test de Shirmer significativement altéré. L'examen du fond d'œil est inappréciable. L'interrogatoire révčle par ailleurs l'absence de larmes męme lors des crises de pleurs, une dysphagie paradoxale qui s'est aggravée avec un retard staturo-pondéral. La réalisation d'un transit œsogastroduodénal combiné ŕ une fibroscopie œsogastroduodénale a montré un rétrécissement régulier du bas œsophage, facilement franchissable sans reflux ni signe d'œsophagite (Figure 2). Le bilan surrénalien est normal.
Observation 2
Fille âgée de 7 ans, 3čme d'une fratrie de 4, issue d'un mariage consanguin présente une baisse d'acuité visuelle bilatérale chiffrée ŕ 2/10čme non améliorable au niveau de l'œil droit et ŕ mouvement des doigts au niveau de l'œil gauche. L'examen au biomicroscope révčle une kératite ponctuée superficielle au niveau de l'œil droit (Figure 3) et une taie de cornée centrale de l'œil gauche avec néovascularisation (Figure 4). L'examen du fond de l'œil est normal ŕ droite. Par ailleurs, ses parents rapportent la notion de vomissements alimentaires puis des épisodes de dysphagie paradoxale. L'examen biologique trouve des taux normaux de cortisol (453µm/l) et d'ACTH (27pg/ml). Le transit œsogastroduodénal est en faveur d'un mégaœsophage (Figure 5) et la fibroscopie œsogastroduodénale en faveur d'une œsophagite stade 1.
Observation 3
Fille âgée de 5 ans, 6čme d'une fratrie de six, issue d'un mariage consanguin chez qui les parents ont constaté l'apparition d'une taie cornéenne de l'OG (Figure 6) et une Alacrymie bilatérale. L'acuité visuelle est estimée ŕ 2/10čme au niveau de l'œil droit, et ŕ mouvement des doigts au niveau de l'œil gauche. L'examen au biomicroscope met en évidence une kératite bilatérale, plus importante ŕ gauche, prenant la fluorescéine et un fond d'œil d'aspect normal. L'interrogatoire révčle la présence de vomissements et une dysphagie paradoxale avec un retard staturopondéral. Le bilan biologique est normal et le TOGD combiné ŕ une FOGD met en évidence un mégaoesophage sans signe d'œsophagite.
Le premier patient a bénéficié d'un traitement ŕ base de larmes artificielles ŕ instillations fréquentes et réguličres et l'achalasie de l'œsophage a été traitée chirurgicalement par voie cœlioscopique: cardiomyotomie de Heller associée ŕ un geste anti-reflux avec de simples suites opératoires. Le deuxičme enfant a bénéficié d'un traitement ŕ base de larmes artificielles en attendant la greffe de cornée, et d'un geste chirurgical identique au précédent avec des suites simples. Le troisičme enfant a bénéficié d'un traitement chirurgical du mégaoesophage par voie cœ'lioscopique avec suppléance lacrymale en insistant sur la nécessité d'une instillation continue. Le recul au delŕ de 6 mois en moyenne est marqué par la persistance de la KPS malgré un traitement assidu et l'absence d'amélioration de l'acuité visuelle. Sur le plan général, les signes digestifs ont nettement régressé en post opératoire avec une légčre amélioration de la courbe de croissance staturo-pondérale. L'insuffisance cortico-surrénalienne n'est, quand ŕ elle, pas apparue jusqu'ŕ ce jour.
Le syndrome d'Allgrove est une affection rare (97 cas publiés), de transmission autosomique récessive, Le gčne responsable de la maladie, localisé sur le chromosome 12, coderait la protéine ALADIN (pour alacrima-achalasia-adrenal insufficiency neurologic disorder), qui appartient ŕ la famille des protéines de régulation. Le rôle exact de cette protéine dans le syndrome Triple A n'est pas encore connu. Mais les chercheurs pensent qu'elle pourrait avoir un rôle de régulateur sur les récepteurs de l'hormone produite par les glandes surrénales, le cortisol, et un rôle dégénératif sur le systčme nerveux [1,2].
En 1978, Allgrove et all rapportent pour la premičre fois le cas de deux frčres et deux sœurs, issus d'un mariage consanguin qui présentent une achalasie, une déficience de l'axe glucocorticoďde, et un défaut de sécrétion lacrymale, leur permettant de définir cette combinaison comme étant un trouble héréditaire familial connu sous le nom du syndrome d'Allgrove ou syndrome des 3 A [3, 4]. Dans les années suivantes, un certain nombre d'auteurs ont publié des rapports similaires qui ont contribué ŕ définir les principaux caractéristiques de ce syndrome [3,4].
L'incidence de la pathologie est inconnue et difficile ŕ déterminer ŕ cause de la variante clinique de la maladie et de la mortalité infantile due aux crises d'insuffisance surrénale, d'oů l'importance d'une anamnčse soigneuse ŕ la recherche d'antécédents de mortalité dans la fratrie. L'Allgrove touche aussi bien le sexe masculin que le sexe féminin sans différence de race (sexe rationnel= 1). L'âge de début est imprécis; l'achalasie et l'alacrymie représentent les signes inaugurateurs de la pathologie et peuvent apparaitre dčs la naissance alors que l'insuffisance surrénale se développe au cours des deux premičres décades de la vie [5]. Par ailleurs, d'autres auteurs ont suggéré le nom du syndrome des 4A: insuffisance surrénale, achalasie, alacrymie, anomalies du systčme autonome, mais tous ont conclu que l'alacrymie représente le signe inaugural de l'affection, suivi de l'achalasie [4-7]. Une association entre le syndrome de triple A et une neutropénie chronique asymptomatique a également été rapporté [3].
Les principales manifestations ophtalmologiques dans le syndrome triple A incluent l'alacrymie avec l'atrophie des glandes lacrymales et absence de larmoiement, les kératoconjontivites, des anomalies pupillaires notamment une lenteur du jeu pupillaire, des troubles de l'accommodation, une amblyopie et une atrophie optique [2,8].
Les processus d'accommodation, de myosis et de larmoiement (basal et reflexe) sont sous contrôle parasympathique et cholinergique, ce qui explique que les troubles observés chez les patients souffrant du syndrome triple A résultent d'un dysfonctionnement du systčme autonome probablement en rapport avec une atteinte centrale [5, 7, 9-11]. Mullaney et al. [8] ont noté la présence de glandes lacrymales atrophiées et une réduction des glandes séreuses sur pičce de biopsie des glandes lacrymales chez trois de leurs patients. Les crises d'insuffisance surrénale peuvent ętre découvertes ŕ l'âge adulte, lors de la 2čme décade, ou peuvent ne jamais apparaître [4, 5], et c'est le cas de nos trois patients. Cette insuffisance peut compromettre le pronostic vital et doit ętre recherchée soigneusement.
D'autres symptômes ont été constatés au cours de la maladie incluant : o Microcéphalie en rapport avec la récurrence des crises d'hypoglycémie et/ou la malnutrition, parfois une dégénérescence neurologique avec retard mental. o Hyperpigmentation, hyperkératose, fissuration de la paume des mains et de la plante des pieds. o Hypotension orthostatique en rapport avec la défaillance du systčme nerveux autonome. o Bronchopneumopathies ŕ répétition due ŕ la dysphagie et l'achalasie.
Le traitement de la maladie d'Allgrove comporte 3 volets: l'alacrymie: application réguličre de lubrifiants et de larmes artificielles; l'achalasie: traitement chirurgical exclusivement (cardiomyotomie de Heller avec geste antireflux) avec un traitement antireflux en post opératoire immédiat afin d'éviter les complications bronchopulmonaires; l'insuffisance surrénale: apport en glucocorticoďdes avec surveillance stricte en milieu hospitalier au début sans omettre le traitement adjuvant (potassium, calcium, vitamine D 3) et un régime hyposodé hypoglycémique. Ce type de pathologie nécessite une surveillance et un suivi multidisciplinaire: ophtalmologique, pédiatrique, neurologique et une éducation du patient sur son hygične de vie quelque soit son âge.
Le syndrome d'Allgrove ou triple A syndrome est un trouble pédiatrique rare, associant alacrymie et achalasie qui sont constants et précoces, et une insuffisance surrénale moins constante. Ces troubles sont ŕ l'origine d'une altération de la qualité de vie des patients imposant une prise en charge multidisciplinaire et surtout un conseil génétique dans la fratrie.
Les auteurs ne déclarent aucun conflit d'intéręts.
Rajae Derrar: examen clinique et suivi des patients, analyse des examens complémentaires, regroupement des données, rédaction de l'article et soumission de l'article. Nourreddine Boutimzine: inspection des examens cliniques, contrôle de l'article avant soumission. Amina Laghmari: inspection des examens cliniques, contrôle de l'article avant soumission. Amal Alouane: Examen clinique et suivi des patients. Rajae Daoudi: contrôle de l'article avant soumission.
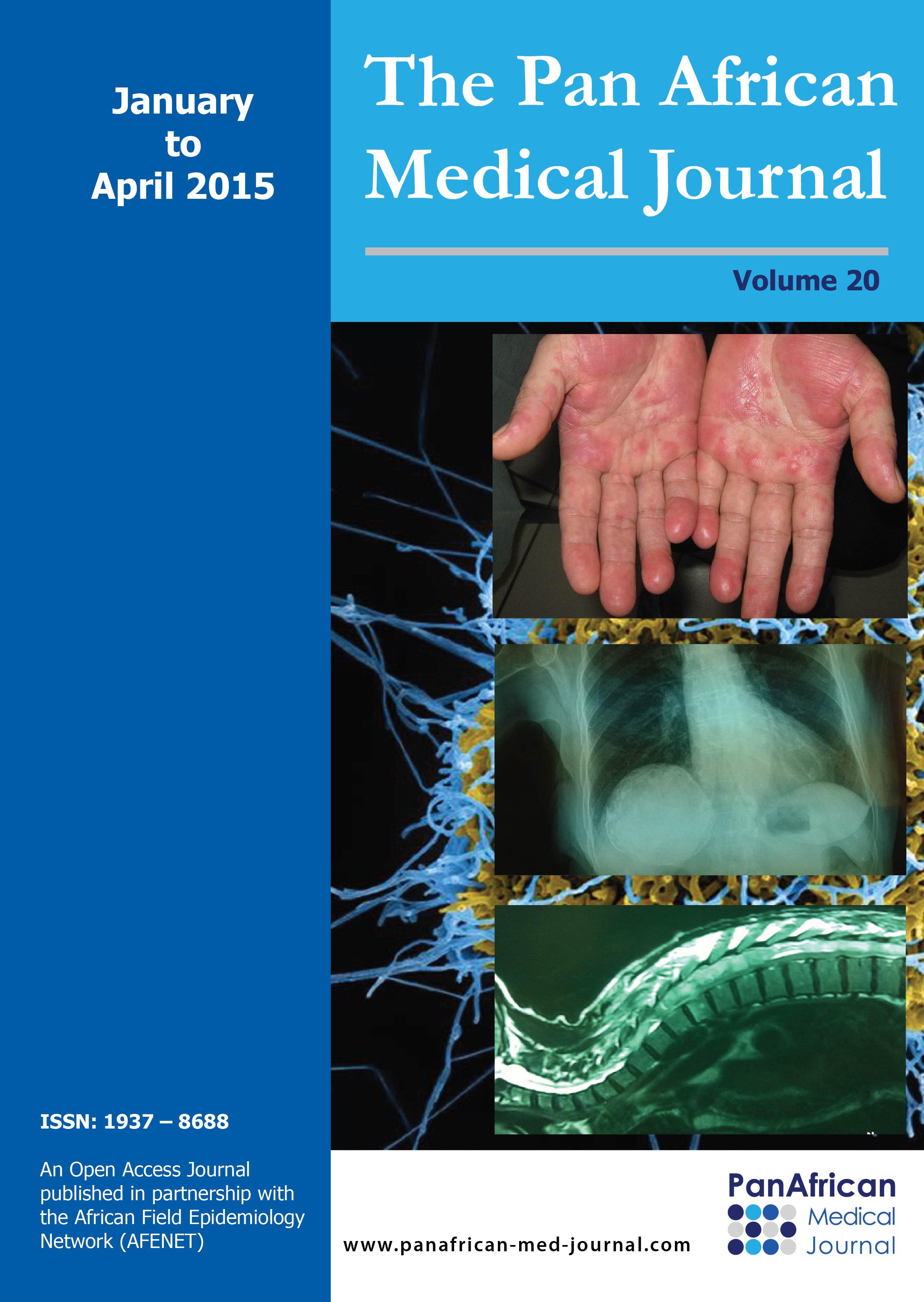
Figure 1: aspect de kératopathie sčche chez un enfant de 4 ans révélé par un test ŕ la fluorescéine
Figure 2: transit śsogastroduodénal montrant un rétrécissement régulier du bas śsophage
Figure 3: kératite ponctuée superficielle au niveau de l’śil droit chez une enfant de 7 ans
Figure 4: taie de cornée centrale de l’śil gauche avec néovascularisation
Figure 5: transit śsogastroduodénal en faveur d’un méga śsophage
Figure 6: taie de cornée de l’śil gauche chez une enfant de 5 ans
- Djemai M, Abada-Bendib M, Masmoudi A-N, Azzôug, Chentli M. Syndrome d'Allgrove : revue de 3 familles. Revue Neurologique. 2007 April;163 (4): 112. PubMed | Google Scholar
- Brooks BP, Kleta R, Caruso RC, Stuart C, Ludlow J, Stratakis CA. Triple A syndrome with prominent ophthalmic features and a novel mutation in the AAAS gene:a case report. BMC Ophthalmol.2004;4:7. PubMed | Google Scholar
- Spiegel R, Shaley S, Huebner A, Horovitz Y. Association of chronic symptomatic neutropenia with the Triple A syndrome. J Pediatr Hematol Oncol. 2005 Jan;27(1):53-5. PubMed | Google Scholar
- Aghajanzadeh M, Safarpoor F, Hydayati MH, Kohssari MR, Mashhour MY, Soleymani AS. Allgrove syndrome: reports of cases and literature. Saudi J Gastroenterol. 2006 Jan-Mar;12(1):34-5. PubMed | Google Scholar
- Kimber J, McLean BN, Prevett M, Hammans SR. Allgrove or 4 "A" syndrome: an autosomal recessive syndrome causing multisystem neurological disease. J Neurol Neurosurg Psychiatry. 2003 May;74(5):654-7. PubMed | Google Scholar
- Gazarian M, Cowell CT, Bonney M, Grigor WG. The "4A" syndrome: adrenocortical insufficiency associated with achalasia, alacrima, autonomic and other neurological abnormalities. Eur J Pediatr. 1995 Jan;154(1):18-23. PubMed | Google Scholar
- Moore PS, Couch RM, Perry YS, Shuckett EP, Winter JS. Allgrove syndrome: An autosomal recessive syndrome of ACTH insensitivity, achalasia and alacrima. Clin Endocrinol (Oxf). 1991;34(2):107-14. PubMed | Google Scholar
- Mullaney P et al . Keratoconjunctivitis sicca associated with achalasia of the cardia, adrenalcortical insufficiency and lacrimal gland degeneration: Keratoconjunctivitis sicca secondary to lacrimal gland degeneration may parallel degenerative changes in esophageal and adrenocortical function. Ophthalmology. 1998;105(4):643-50. PubMed | Google Scholar
- Kalpana Babu, Krishna R Murthy, Narendra Babu, S Ramesh.Triple A syndrome with ophthalmic manifestations in two siblings. Indian journal of oph. 2007; 55(4):304-6. PubMed | Google Scholar
- Wanders RJ, Van Roermun CW, Schelen, A, Schutgens RBH, Zeman J, Kozich V, Hyanek J, Casteels M, Mannaerts GP.Di-and trihydroxycholestanaemia in twin sisters. J Inherit Metab Dis. 1991;14(3): 357-60. PubMed | Google Scholar
- Heinrichs C, Tsigos C, Descheppe RJ, Drews R, Collu R, Dugardeyn C, Goyens P, Ghanem GE, Bosson D, Chrousos GP et al. Familial adrenocorticotropin unresponsiveness associated with alacrima and achalasia: biochemical and molecular studies in two siblings with clinical heterogeneity. Eur J Pediatr. 1995;154(3):191-6. PubMed | Google Scholar
Search
This article authors
On Pubmed
On Google Scholar
Citation [Download]
Navigate this article
Similar articles in
Key words
Tables and figures






Article metrics
Recently from the PAMJ








Authors´ services
