
Sight threatening Vitreous Haemorrhage and retinal detachment in a patient with sickle cell disease
Jemal Zeberga Shifa, Alemayehu Mekonnen Gezmu
Corresponding author: Jemal Zeberga Shifa, Department of Surgery, Medical Faculty, University of Botswana, Botswana 
Received: 14 Sep 2018 - Accepted: 17 Dec 2019 - Published: 02 Jan 2020
Domain: Ophthalmology
Keywords: Sickle cell disease, vitreous haemorrhage, retinal detachment
©Jemal Zeberga Shifa et al. Pan African Medical Journal (ISSN: 1937-8688). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: Jemal Zeberga Shifa et al. Sight threatening Vitreous Haemorrhage and retinal detachment in a patient with sickle cell disease. Pan African Medical Journal. 2020;35:1. [doi: 10.11604/pamj.2020.35.1.17098]
Available online at: https://www.panafrican-med-journal.com//content/article/35/1/full
Case report 

Sight threatening Vitreous Haemorrhage and retinal detachment in a patient with sickle cell disease
Sight threatening vitreous haemorrhage and retinal detachment in a patient with sickle cell disease
Jemal Zeberga Shifa1,&, Alemayehu Mekonnen Gezmu2
1Department of Surgery, Medical Faculty, University of Botswana, Botswana, 2Department of Paediatrics and Adolescent Health, Faculty of Medicine, University of Botswana, Botswana
&Corresponding author
Jemal Zeberga Shifa, Department of Surgery, Medical Faculty, University of Botswana, Botswana
We report a case of sight threatening vitreous haemorrhage and retinal detachment as complication of sickle cell disease (SCD). A 35 years old female Nigerian patient had presented to ophthalmology clinic of Princess Marina Hospital, Botswana, with two weeks history of poor vision in the left eye. The loss of vision was due to vitreous haemorrhage and retinal detachment which was confirmed by direct and indirect ophthalmoscopy and B-Scan ultrasound. Prior to presentation, patient didn't have any follow up by an ophthalmologist as part of regular medical care for patients with SCD. We emphasize the importance of regular follow up for early detection, treatment and prevention of complication associated with sickle cell disease.

Normal red blood cell haemoglobin comprises four polypeptide globin chains, each associated with a central hemering (ferroprotoporphyrin). The globin chains consist of two identical pairs of α and β polypeptide chains. The sickle-cell haemoglobinopathies are characterized by a genetic error in β-chain synthesis, which results in abnormal function of the haemoglobin molecule. In conditions of ischemia and metabolic stress, the imperfect globin chains induce pathologic alterations in red blood cell morphology. Owing to their crescent shape, the altered red blood cells are labelled ´sickle cells´ [1]. The phenotypic heterogeneity of sickle cell disease (SCD) ranges from haemolytic anaemia and chronic vasculopathy with acute painful crises to a multitude of organ-specific manifestations of varying severity. The exact cause of this diversity of clinical features remains unclear, but recent advances in the understanding of clinical SCD indicate that the pathophysiology of this disease is complex and multifaceted. Factors that influence the disease process include genetic variation, the proportion of sickled cells, endothelial cell activation, inflammation, oxidative stress and hypoxia-induced angiogenesis [2,3]. Since many of the problems are time dependent, the overall prevalence of ocular complications of sickle disease is unknown. Although various systemic complications of SCD are known to be more common in patients with the Hb SS genotype, visual impairment secondary to proliferative sickle retinopathy is more common in patients with the Hb SC genotype. In a large cohort, 6% (49/783) of Hb SS genotype patients had proliferative sickle retinopathy (PSR) on initial examination compared with 32% (172/533) of Hb SC genotype patients [4]. Approximately 40-43% of haemoglobin Hb SC genotype patients and 14-20% of haemoglobin Hb SS patients were expected to develop PSR [5, 6].

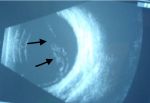
A 35 years old female Nigerian patient was diagnosed to have Sickle cell disease since early child hood. She presented to our ophthalmology clinic, in Gaborone, Botswana, with reduction of vision in the left eye for two weeks. During initial evaluation, the visual acuity in right eye was 6/6 and in the left eye counting fingers from a distance of 5 meters. Intraocular pressure in both eyes was with in normal range. Posterior segment evaluation using direct and indirect ophthalmoscopy showed retinal detachment and vitreous haemorrhage in the left eye. B scan ultrasound showed vitreous opacity with retinal detachment in the left eye (Figure 1). The patient was followed in our clinic for more than 3 months with no improvement in her vision on the affected side. Decision was made to perform parsplana vitrectomy. However, the patient refused the surgery despite repeated counselling. Currently her vision on left eye is only light perception.
Nearly all ocular or periocular structures can be affected by the sickle haemoglobinopathies. Classic anterior segment findings include conjunctival ´comma-shaped´ capillaries which represent intravascular sludging of sickling red blood cells [7]. Sectorial iris atrophy represents areas of anterior uveal ischemia. A mild anterior chamber cell and flare reaction may be observed secondary to incompetence of the blood-ocular barrier. The anterior segment manifestations generally do not pose significant risks for vision loss. Posterior segment manifestations of sickle haemoglobinopathies may be observed in the vitreous body, optic disc, retina and sub retinal structures. Vitreous haemorrhage secondary to peripheral retinal neovascularization may develop. The optic disc may demonstrate sludging red blood cells within prepapillary retinal capillaries. More debilitating macular ischemia can result in frank macular infarction with an enlarged foveal avascular zone secondary to multiple retinal arteriolar occlusions with an often insidious, progressive course [8,9]. Our patient presented with advanced stage of ocular complication of SCD because of lack of awareness about the importance of regular ophthalmologic evaluation by the patient and allied health care professional for early identification of ocular complications of SCD.
We emphasize the importance of ophthalmologic regular check-up for patients with SCD. Delay in diagnosis and management can lead to loss of vision as the result of ocular complication of sickle cell disease.
The authors have no competing interests.
JZS managed the patient, wrote the first draft of the manuscript. AMG revised the manuscript and checked for consistency.
Figure 1: B scan ultrasound of the left eye with evidence of vitreous haemorrhage and retinal detachment (arrows)
- Hoffman R, Furie B, Benz J. Haematology: Basic Principles and Practice. 2009. Philadelphia. Churchill Livingstone.
- Elagouz M, Jyothi S, Gupta B, Sivaprasad S. Sickle cell disease and the eye: old and new concepts. Surv Ophthalmol. 2010;55(4):359-77. PubMed | Google Scholar
- Garg S, Jampol LM, Rabb M. Sickle-cell disease. Duane´s Clinical Ophthalmology. 2009. Philadelphia. Lippincott, Williams & Wilkins.
- Fadugbagbe AO, Gurgel RQ, Mendonça CQ, Cipolotti R, Dos Santos AM, Cuevas LE. Ocular manifestations of sickle cell disease. Ann Trop Paediatr. 2010 Mar 1;30(1):19-26. PubMed | Google Scholar
- Condon PI, Serjeant GR. Behaviour of untreated proliferative sickle retinopathy. Br J Ophthalmol. 1980 Jun 1;64(6):404-11. PubMed | Google Scholar
- Paton D. The conjunctival sign of sickle-cell disease. Arch Ophthalmol. 1961 Jul 1;66:90-4. PubMed | Google Scholar
- Asdourian GK, Goldberg MF, Rabb MF. Macular infarction in sickle cell B+ thalassemia. Retina. 1982;2(3):155-8. PubMed | Google Scholar
- Merritt JC, Risco JM, Pantell JP. Bilateral macular infarction in SS disease. J Pediatr Ophthalmol Strabismus. 1982;19(5):275-8. PubMed | Google Scholar
- Sanders RJ, Brown GC, Rosenstein RB, Magargal L. Foveal avascular zone diameter and sickle cell disease. Arch Ophthalmol. 1991 Jun 1;109(6):812-5. PubMed | Google Scholar
Search
This article authors
On Pubmed
On Google Scholar
Citation [Download]
Navigate this article
Similar articles in
Key words
Tables and figures
Article metrics
Recently from the PAMJ








Authors´ services
