
Le corticosurrénalome: une cause exceptionnelle d’hyperaldostéronisme primaire
Baha Zantour, Ines Charrada, Zohra Elati, Fatma Larbi Ammari, Fadia Boubaker, Sondes Arfa, Olfa Berriche, Wafa Alaya, Mohamed Habib Sfar
Corresponding author: Baha Zantour, Service d’Endocrinologie et Médecine Interne, Hôpital Tahar Sfar, Mahdia, Tunisie 
Received: 01 Sep 2018 - Accepted: 15 Sep 2018 - Published: 27 Sep 2018
Domain: Endocrinology
Keywords: Corticosurrénalome malin, hyperaldostéronisme primaire, surrénale
©Baha Zantour et al. Pan African Medical Journal (ISSN: 1937-8688). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: Baha Zantour et al. Le corticosurrénalome: une cause exceptionnelle d’hyperaldostéronisme primaire. Pan African Medical Journal. 2018;31:60. [doi: 10.11604/pamj.2018.31.60.16973]
Available online at: https://www.panafrican-med-journal.com//content/article/31/60/full
Original article 

Le corticosurrénalome: une cause exceptionnelle d’hyperaldostéronisme primaire
Le corticosurrénalome: une cause exceptionnelle d’hyperaldostéronisme primaire
Corticosurrenaloma: an exceptional cause of primary hyperaldosteronism
Baha Zantour1,&, Ines Charrada1, Zohra Elati1, Fatma Larbi Ammari1, Fadia Boubaker1, Sondes Arfa1, Olfa Berriche1, Wafa Alaya1, Mohamed Habib Sfar1
1Service d’Endocrinologie et Médecine Interne, Hôpital Tahar Sfar, Mahdia, Tunisie
&Auteur correspondant
Baha Zantour, Service d’Endocrinologie et Médecine Interne, Hôpital Tahar Sfar, Mahdia, Tunisie
Le corticosurrénalome est une tumeur maligne rare de la cortico-surrénale. Il secrčte souvent des corticostéroďdes, des stéroďdes sexuels et des précurseurs. Le corticosurrénalome producteur d'aldostérone est trčs rare, 1cas/10 millions d'habitants. Nous rapportons l'observation d'un homme de 38 ans se présentant pour hypertension artérielle sévčre associée ŕ une hypokaliémie profonde (2.2 mmol/l). L'exploration a conclu ŕ un hyperaldostéronisme primaire (aldostérone = 2645pmol/l, rapport aldostérone/rénine = 327pmol/mUI), avec hypersécrétion de glucocorticoďdes. Le scanner abdomino-pelvien a montré une masse surrénalienne gauche de 9cm, mal limitée et hétérogčne, infiltrant la graisse autour et le diaphragme, envahissant la veine rénale gauche, avec adénopathie régionale et nodule hépatique de 4 cm. Le patient a eu une néphrectomie élargie, suivie d'une hépatectomie droite deux mois aprčs entrainant une rémission. Un an aprčs le patient a développé des métastases pulmonaires. Nous concluons que l'hyperaldostéronisme primaire peut ętre un mode de révélation du corticosurrénalome, on devra y penser malgré son caractčre exceptionnel.
English abstract
Corticosurrenaloma is a rare malignant tumor of the adrenal gland that often secretes corticosteroids, sex steroids and precursors. Aldosterone-producing corticosurrenaloma is very rare, accounting for 1 case/10million inhabitants. We report the case of a 38-year old man presenting with severe arterial hypertension associated with deep hypokalaemia (2.2 mmol/L). Exploration showed primary hyperaldosteronism (aldosterone = 2645 pmol/l, aldosterone/renin ratio = 327 pmol/MUI), with hypersecretion of glucocorticoids. Abdominopelvic CT scan revealed left poorly differentiated and heterogeneous adrenal mass measuring 9cm, infiltrating into the surrounding adipose tissue and the diaphragm, extending into the left renal vein, with regional adenopathy and hepatic nodule measuring 4cm. The patient underwent radical nephrectomy followed by right hepatectomy two months after resulting in remission. A year after the patient developed lung metastases. This study highlights that corticosurrenaloma should be suspected in patients with primary aldosteronism despite its rarity.
Key words: Malignant corticosurrenaloma, primary hyperaldosteronism, adrenal

Le corticosurrénalome (CS) est une tumeur endocrine maligne rare de la corticosurrénale. Son incidence est d'environ 1 ŕ deux nouveaux cas par million et par an [1]. Le plus fréquemment, le diagnostic est posé devant des signes d'hypersécrétion hormonale dont la symptomatologie est variable, dépendant de la voie de synthčse hormonale développée par le tissu endocrinien tumoral et/ou devant un syndrome tumoral. Dans tous les cas, une altération de l'état général est trčs souvent notée et le mode de révélation est trčs rapide [2]. Le CS producteur d'aldostérone est trčs rare, un cas par dix millions d'habitants [3]. Nous rapportons ici un cas de CS secrétant de l'aldostérone et des gluco-corticoďdes, découvert devant une HTA et une hypokaliémie sévčres isolées, sans altération de l'état général ni de syndrome tumoral ni de signes cliniques en faveur de la sécrétion gluco-corticoide.

Il s'agit d'un homme âgé de 37 ans, commerçant, marié, pčre d'une fille, aux antécédents familiaux d'hypertension artérielle chez la mčre. Son histoire remonte ŕ un mois auparavant, soit le 13/01/17 marquée par la survenue de lipothymie amenant le malade ŕ consulter les urgences oů une pression artérielle ŕ 190/85 mm Hg a été objectivée. Il a été mis initialement sous Nicardipine LP 50mg deux fois par jour. A l'interrogatoire, le patient rapportait des céphalées, des crampes musculaires, une hypersudation et une prise de poids. A l'examen clinique, Il n'avait pas de vergetures ni d'œdčmes des membres inférieurs, poids 78kg, taille 1.65m, indice de masse corporelle 28,67 kg/m2. Le bilan biologique a révélé une hypokaliémie profonde ŕ 2,2 mmol/l, hyperkaliurčse ŕ 98,8 mmol/l, TSH 1.44 mUI/l, protéinurie de 24h négative, calcémie: 2,42 mmol/l. Le reste du bilan a montré: clairance de la créatinine 74 ml/min, glycémie 5,8 mmol/l, cholestérol 6,5 mmol/l, triglycérides 1,38 mmol/l, HDL cholestérol 1,53 mmol/l et LDL cholestérol 4,3 mmol/l. L'exploration hormonale en conditions standardisées [4] (patient en position assise depuis 15 min et aprčs 2 heures de réveil) a montré: Aldostéronémie 2645 pmol/l, Rénine 8,1 mUI/l, Rapport aldostérone/rénine 327 pmol/mUI. L'HAP a été ainsi confirmé devant une aldostéronémie largement supérieure au seuil de 550 pmol/l, une rénine basse < 5 mUI/l et un rapport supérieur ŕ 64. L'échographie abdominale a montré une masse surrénalienne gauche finement calcifiée dépassant 4 cm de grand axe, un nodule hypoéchogčne du segment VIII du foie de 36 mm de grand axe, des micro lithiases rénales bilatérales sans retentissement.
Le scanner abdomino-pelvien fait le 05/02/17 a montré une masse surrénalienne gauche de 9cm, mal limitée et hétérogčne avec hémorragie, nécrose et calcifications, infiltration de la graisse autour et du diaphragme, invasion de la veine rénale gauche sans extension ŕ la veine cave, adénopathie régionale (Figure 1, Figure 2), et nodule secondaire hépatique de 4cm (Figure 3). Cet aspect était en faveur d'un corticosurrénalome stade IV de l'ENSAT [5] nécessitant la poursuite des explorations par un bilan hormonal et d'extension Explorations hormonales: cortisolémie 8h 175,8 µg/l (> 130 µg/l), cortisolémie 20h 188,6 µg/l (> 75 µg/l), cortisol libre urinaire CLU 192 µg/24h (> 80µg/24H). L'élévation du cortisol plasmatique et urinaire et l'inversion du cycle du cortisol étaient trčs en faveur d'un hypercorticisme qui a été confirmé par l'absence de freinage au test de freinage faible par la dexamethazone (2mg/jour pendant 2 jours): Cortisolémie: 185,4 microg/l, CLU: 142.5 microg/24h. Le dosage de l'ACTH a trouvé une valeur freinée ŕ 3,9 pg/ml. Il s'agissait donc d'un syndrome de Cushing ACTH indépendant. Il n'y avait pas d'excčs de production des stéroďdes sexuels, œstradiol: 25 ng/ml, testostérone plasmatique: 2.52 ng/ml. Le dosage des dérivés méthoxylés urinaires des 24h était normal. A l'issue de cet ensemble d'investigations convergentes, le diagnostic retenu est celui d'un CS stade IV avec invasion loco-régionale, métastase hépatique, et extension ŕ la veine rénale gauche, hypersécrétant d'aldostérone et de glucocorticoďdes. Le patient a été mis sous spironolactone 100 mg un comprimé par jour et chlorure de potassium 600 mg, un comprimé trois fois par jour. Il a eu une néphrectomie élargie, suivie d'une hépatectomie droite deux mois aprčs entrainant une rémission. Un an aprčs, soit en mars 2018, le patient a développé des métastases pulmonaires qui ont été traitées par radiothérapie externe. Le test au Tetracosactide (ACTH synthétique) 1 microg fait en mai 2018 a montré une insuffisance surrénalienne. Le patient est actuellement sous hydrocortisone 10 mg un comprimé par jour, sa pression artérielle et kaliémie sont normales, a perdu 10kg de poids. Un traitement par mitotane est prévu.
Le CS est une tumeur maligne rare dont le tableau clinique est variable. Les CS fonctionnels ou sécrétants entraînent un syndrome endocrinien. Par ordre de fréquence, il peut s'agir d'un hypercorticisme isolé ou associé ŕ une virilisation ou une féminisation [1, 6], d'une hyperandrogénie pouvant provoquer chez le nouveau-né une ambiguité sexuelle ŕ la naissance [7] et exceptionnellement une hyperoestrogénie ou un HAP [8], c'est le cas de notre patient. L'HAP peut ętre un mode de révélation d'un CS [9]. Il est responsable d'un tableau clinico-biologique dont la sévérité peut ętre en rapport avec la nature carcinomateuse de la lésion. La littérature confirme la rareté de l'exclusivité de la production d'aldostérone par le CS [9]. La TDM abdominale est l'examen de premičre intention pour l'exploration des surrénales devant un syndrome d'hyper-sécrétion hormonale [4]. L'existence d'une hémorragie intra-tumorale ou d'une nécrose donne un aspect scannographique hétérogčne en faveur de la malignité sans que ce soit spécifique [10]. Les adénomes de Conn sont souvent inférieurs ŕ 5cm, tandis que les carcinomes sont souvent plus larges [3]. A notre connaissance, 64 cas de CS entrainant un HAP ont été rapportés [7, 9, 11-15]. Seccia TM et al. ont fait une revue de 58 cas de CS produisant l'aldostérone [2]. Selon cette revue, l'hypokaliémie était présente dans tous les cas sauf un seul cas [8], l'hypertension artérielle a été trouvée dans tous les cas sauf 5 cas [2, 12]. Plusieurs hypothčses ont été avancées pour expliquer l'absence d'hypertension artérielle, mais le mécanisme exact est inconnu. Dans 10% des cas il y avait une augmentation modérée du cortisol associée et la S-DHEA était souvent normale [2]. Les taux des 17-hydroxystéroides et des 17-cétosteroides, mesurés dans 28 cas, étaient augmentés dans 8% et 6% des cas respectivement.
Le traitement de premičre intention des CS quel que soit le type de sécrétion est la chirurgie résultant en une rémission dans 0 ŕ 50% des cas selon le stade lors de l'intervention [16]. La survie moyenne est de 4 ŕ 8 mois lorsque les localisations secondaires existent [17]. Chez notre patient, la chirurgie a entrainé une rémission de un an avec normalisation de la pression artérielle et de la kaliémie et insuffisance cortisolique malgré la présence de métastase hépatique et d'une extension ŕ la veine rénale. La chirurgie surrénalienne doit ętre proposée systématiquement dans les CS męme en présence de métastases si une résection complčte peut ętre obtenue [18]. De plus, la chirurgie des métastases sera envisagée, en cas de facteurs pronostiques favorables, lorsque la résection peut ętre complčte et ŕ chaque fois qu'une réponse tumorale ŕ la chimiothérapie est observée [5]. Chez notre patient, la chirurgie surrénalienne et hépatique ont pu allonger la survie et permis de maitriser l'hyper-sécrétion hormonale. L'alternative aux ré-interventions reste le mitotane et les polychimiothérapies. En cas de récidive oů l'exérčse a été incomplčte ou récusée, le traitement de premičre ligne est le mitotane. Le traitement par mitotane doit ętre adapté au taux sérique qui doit ętre supérieur ŕ 14 μg/ml pour atteindre une dose thérapeutique efficace [14]. Le mitotanne associé ŕ la chimiothérapie est plus efficace que le mitotane seul. L'association mitotane/étoposide/doxorubicine/cisplatine a été démontrée supérieure ŕ l'association mitotane/streptozotocine dans les formes non résécables et/ou métastatiques dans l'étude FIRM-ACT [19]. La radiothérapie adjuvante du lit tumoral n'a pas d'effet favorable démontré. Par contre, la radiothérapie hépatique, pulmonaire ou osseuse peut ętre utilisée notamment sur les localisations hépatiques et pulmonaires lorsque celles-ci sont peu nombreuses et d'agressivité intermédiaire [19]. Une radiothérapie thoracique des métastases pulmonaires a été pratiquée chez notre patient, avec un recul actuel de 6 mois.
Le CS est une tumeur maligne rare. Le diagnostic a été établi dans notre cas sur des critčres cliniques, biologiques et radiologiques malgré le caractčre exceptionnel et trompeur de son mode de révélation qui était l'hyperaldostéronisme primaire. La prise en charge nécessite une collaboration multidisciplinaire qui a été mise en œuvre pour notre patient. L'évolution est imprévisible. Chez notre patient, la chirurgie de la tumeur primitive et de la métastase hépatique puis la radiothérapie des métastases pulmonaires a permis d'allonger la survie et de traiter efficacement l'hyper-sécrétion hormonale. L'étude génétique de la pičce tumorale qui a manqué dans notre observation aurait eu l'intéręt d'estimer le pronostic et l'évolution.
Les auteurs ne déclarent aucun conflit d'intéręts.
Chacun des auteurs a participé ŕ la prise en charge du patient du diagnostic au traitement. Les 2 premiers auteurs ont réalisé la rédaction de l'article. Tous les auteurs ont lu et approuvé la version finale du manuscrit.
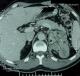
Figure 1: TDM abdominale:
coupe transversale: corticosurrénalome gauche de 9 cm; les flčches montrent
les calcifications et les nécroses
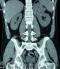
Figure 2: TDM abdominale:
coupe sagittale de la tumeur
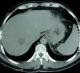
Figure 3: TDM abdominale:
métastase hépatique droite de 4 cm
- Allolio B, Fassnacht M. Adrenocortical Carcinoma: clinical update. J Clin Endocrinol Metab. 2006 Jun; 91(6): 2027-37. Epub 2006 Mar 21. PubMed
- Seccia TM, Fassina A, Nussdorfer GG, Pessina AC et al. Aldosterone-producing adrenocortical carcinoma: an unusual cause of Conn's syndrome with an ominous clinical course. Endocr Relat Cancer. 2005 Mar; 12(1): 149-159. PubMed | Google Scholar
- Weingartner K, Gerharz EW, Bittinger A, Rosai J. Isolated clinical syndrome of primary aldosteronism in a patient with adrenocortical carcinoma. Urol Int. 1995; 55(4): 232-235. PubMed | Google Scholar
- Funder JW, Carey RM, Mantero F, Murad MH et al. The Management of Primary Aldosteronism: Case Detection, Diagnosis and Treatment: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2016 May; 101(5): 1889-1916. PubMed | Google Scholar
- Fassnacht M, Johanssen S, Quinkler M, Bucsky P et al. Limited prognostic value of the 2004 International Union Against Cancer staging classification for adrenocortical carcinoma: proposal for a Revised TNM Classification. Cancer. 2009 Jan; 115(2): 243-250. PubMed | Google Scholar
- Chen QL, Su Z, Li YH, Ma HM et al. Clinical characteristics of adrenocortical tumors in children. J Pediatr Endocrinol Metab. 2011; 24(7-8): 535-541. PubMed | Google Scholar
- Pigeon B, Ryckewaert P, Massin B. Virilizing adrenal cortical tumor in children: a propos of a neonatal case. Pediatrie. 1985 Jun; 40(4): 309-312. PubMed | Google Scholar
- Kidd MT, Karlin NJ, Cook CB. Feminizing adrenal neoplasms: case presentations and review of the literature. J Clin Oncol. 2011 Feb; 29(6): e127-130. PubMed | Google Scholar
- Griffin AC, Kelz R, LiVolsi VA. Aldosterone-secreting adrenal cortical carcinoma: a case report and review of the literature. Endocr Pathol. 2014 Sep; 25(3): 344-349. PubMed | Google Scholar
- Drevet D, Guzelian J, Van Box Som P, Champion M et al. Malignant adrenocortical carcinoma with atypical aspect: value of tomodensitometry and magnetic resonance imaging; a propos of a case. Ann Radiol. 1996; 39(3): 126-130. PubMed | Google Scholar
- Mario MA, Claudia RR, Analleli ML, Pedro PG et al. A Rare Presentation of Primary Hyperparathyroidism with Concurrent Aldosterone-Producing Adrenal Carcinoma. Case Rep Endocrinol. 2015 Jun; 2015: 910984. Google Scholar
- Min Soo S, Sung Woo S, Sang Byung B, Yeo Joo K et al. Aldosterone-Producing Adrenocortical Carcinoma without Hypertension. Korean J Intern Med. 2012 Jun; 27(2): 221-223. PubMed | Google Scholar
- Kennosuke O, Takeshi H, Masaya S, Hiroyuki I et al. Aldosterone-producing adrenocortical carcinoma with prominent hepatic metastasis diagnosed by liver biopsy: a case report. BMC Endocrine Disorders. 2016 Jan; 16: 3. PubMed | Google Scholar
- Lazaro K, Adorable PW. Aldosterone-producing Adrenocortical carcinoma with co-secretion of cortisol and estradiol: case report. Journal of the Asean Federation of Endocrine Societies. 2018 Nov; 33(1): 57-62. Google Scholar
- Hussain S, Panteliou E, Berney DM, Carpenter R et al. Pure aldosterone-secreting adrenocortical carcinoma in a patient with refractory primary hyperaldosteronism. Endocrinol Diabetes Metab Case Rep. 2015 Jul; 2015: 150064. PubMed | Google Scholar
- Baudin E, Pellegriti G, Bonnay M, Penfornis A et al. Impact of monitoring plasma 1,1 dichlorodiphenildichloroethane (o,p'DDD) levels on the treatment of patients with adrenocortical carcinoma. Cancer. 2001 Sep; 92(6): 1385-1592. Google Scholar
- Pommier RF, Brennan MF. An eleven-year experience with adrenocortical carcinoma. Surgery. 1992 Dec; 112(6): 963-971. PubMed | Google Scholar
- Fay AP, Elfiky A, Teló GH, McKay RR et al. Adrenocortical carcinoma: the management of metastatic disease. Crit Rev Oncol Hematol. 2014 Nov; 92(2): 123-132. PubMed | Google Scholar
- Hermsen IGC, Groenen YE, Dercksen MW, Theuws J et al. Response to radiation therapy in adrenocortical carcinoma. J Endocrinol Invest. 2010 Nov; 33(10): 712-714. PubMed | Google Scholar
Search
This article authors
On Pubmed
On Google Scholar
Citation [Download]
Navigate this article
Similar articles in
Key words
Tables and figures
Article metrics
Recently from the PAMJ








Authors´ services
