
Holoprosencéphalie alobaire avec diabčte insipide et hypothyroďdie chez un nourrisson de 10 mois
Ndiogou Seck, Idrissa Basse, Younoussa Keita, Djiril Boiro, Lamine Thiam, Aliou Adoulaye Ndongo, Ibrahima Diagne
Corresponding author: Ndiogou Seck, Service de Pédiatrie, UFR des Sciences de la Santé, Université Gaston Berger de Saint-Louis, Sénégal 
Received: 29 Nov 2016 - Accepted: 06 Dec 2016 - Published: 01 Nov 2017
Domain: Maternal and child health
Keywords: Holoprosencéphalie alobaire, nourrisson, diabète insipide, hypothyroïdie centrale
©Ndiogou Seck et al. Pan African Medical Journal (ISSN: 1937-8688). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: Ndiogou Seck et al. Holoprosencéphalie alobaire avec diabčte insipide et hypothyroďdie chez un nourrisson de 10 mois. Pan African Medical Journal. 2017;28:193. [doi: 10.11604/pamj.2017.28.193.11288]
Available online at: https://www.panafrican-med-journal.com//content/article/28/193/full
Original article 

Holoprosencéphalie alobaire avec diabčte insipide et hypothyroďdie chez un nourrisson de 10 mois
Holoprosencéphalie alobaire avec diabčte insipide et hypothyroďdie chez un nourrisson de 10 mois
Alobar holoprosencephaly associated with diabetes insipidus and hypothyroidism in a 10-month old infant
Ndiogou Seck1,&, Idrissa Basse2, Younoussa Keita3, Djiril Boiro3, Lamine Thiam4, Aliou Adoulaye Ndongo3, Ibrahima Diagne1
1Service de Pédiatrie, UFR des Sciences de la Santé, Université Gaston Berger de Saint-Louis, Sénégal, 2Service de Pédiatrie, UFR des Sciences de la Santé, Université de Thiés, Sénégal, 3Service de Pédiatrie, Faculté de Médecine, de Pharmacie et d’Odonto-stomatologie, Université Cheikh Anta Diop de Dakar (Sénégal), 4Service de Pédiatrie, UFR des Sciences de la Santé, Université Assane Seck de Ziguinchor, Sénégal
&Auteur correspondant
Ndiogou Seck, Service de Pédiatrie, UFR des Sciences de la Santé, Université Gaston Berger de Saint-Louis, Sénégal
L’holoprosencéphalie (HPE) est une malformation cérébrale grave due ŕ un défaut de clivage médian du prosencéphale. C’est une anomalie le plus souvent associée ŕ des malformations cranio-faciales, d’un retard du développement psychomoteur, d’un diabčte insipide et de troubles endocriniens variables. Les étiologies sont diverses regroupant les anomalies chromosomiques (trisomie 13, 18), les syndromes polymalformatifs (syndrome de CHARGE). Le diagnostic repose sur l’imagerie cérébrale. Quelques rares cas ont été décrits dans la littérature. Nous rapportons le cas d’une HPE alobaire chez un nourrisson de 10 mois. Le diagnostic a été posé au scanner cérébral devant la constatation d’un retard du développement psychomoteur en l’absence de malformations visibles. Le bilan endocrinien a permis de déceler un diabčte insipide central et une hypothyroďdie centrale probablement d’origine hypothalamique.
English abstract
Holoprosencephaly (HPE) is a serious brain malformation due to a failure of medial forebrain cleavage. This is an abnormality which is more often associated with craniofacial malformations, psychomotor development delay, diabetes insipidus and variable endocrine disorders. It is due to different causes including chromosomal abnormalities (trisomy 13, 18)and polymalformative syndromes (CHARGE Syndrome). Diagnosis is based on brain imaging. A few rare cases have been described in the literature. We here report the case of alobar HPE in a 10-month old infant. Diagnosis was based on cerebral CT scan performed due to delayed psychomotor development and in the absence of visible malformations. Endocrine assessment allowed to detect central diabetes insipidus and central hypothyroidism, probably of hypothalamic origin.
Key words: Alobar holoprosencephaly, infant, diabetes insipidus, central hypothyroidism

L’holoprosencéphalie (HPE) est une malformation cérébrale grave due ŕ un défaut de clivage médian du prosencéphale survenant ŕ un stade embryonnaire trčs précoce, entre le 18e et le 28e jour de gestation. Trois formes anatomiques de sévérité variables ont été décrites: l’HPE alobaire, semi lobaire et lobaire. Elle peut ętre ŕ l’origine d’anomalies faciales, de manifestations neurologiques et de troubles endocriniens divers. Nous rapportons un cas d’holoprosencéphalie alobaire associé ŕ des troubles endocriniens ŕ type: d’hypothyroďdie et d’hypernatrémie neurogéne chez un nourrisson de 10 mois.

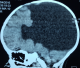
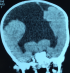
AS, nourrisson de sexe masculin âgé de 10 mois est troisičme d’une fratrie de trois enfants. Il est issu d’un mariage consanguin au deuxičme degré. Sa mčre âgée de 20 ans est sans tare connue. L’accouchement s’est fait par voie basse, le score d’Apgar était de 10 ŕ la 5e minute. Il avait bénéficié d’un allaitement maternel exclusif jusqu’ŕ l’âge de 6 mois puis d’une bonne diversification alimentaire, il était bien vacciné selon le (PEV) programme élargi de vaccination du Sénégal. Sur le plan du développement psychomoteur AS âgé de 10 mois ne pouvait pas se mettre en position assise. Il a été admis dans notre service pour ce retard du développement psychomoteur. A l’admission il avait un retard staturo-pondéral sévčre avec: un poids de 7 kg (inférieur ŕ-3 DS), une taille de 65 cm (inférieur ŕ-3 DS), un périmčtre crânien de 45 cm (entre 0 et-1 DS), une température de 37° Celsius et une diurčse horaire de 3,35 ml/Kg/h. L’examen retrouvait un nourrisson avec une déshydratation modérée (sécheresse des muqueuses, une fontanelle antérieure déprimée et un enfoncement des globes oculaires. Sur le plan neurologique il était conscient, il ne pouvait pas se tenir en position assis par contre la tenue de la tęte était possible. L’examen morphologique ne retrouvait aucune malformation visible et le reste de l’examen était sans particularités. A la biologie: l’ionogramme sanguin retrouvait une hypernatrémie ŕ 173 mmol/l, avec hyper osmolarité plasmatique (351 mosm/l) et l’ionogramme urinaire mettait en évidence une natriurčse basse ŕ 20 mmol/24h et une osmolarité urinaire ŕ 55 mosm/l. Le bilan endocrinien montrait une hypothyroďdie périphérique avec un taux de TSH ŕ 8 mUI/l, la T3 libre ŕ 0,3 nmol/L et la T4 libre ŕ 3 pmol/L. le reste du bilan hormonal (testostéronémie et dosage de l’hormone de croissance) était normal (Tableau 1). A l’imagerie, le scanner cérébral mettait en évidence une holoprosencéphalie alobaire (Figure 1, Figure 2), l’hypophyse n’a pu ętre explorée. Les échographies abdominale et cardiaque étaient normales. La thyroďde était visible sans dysmorphie ŕ l’échographie cervicale. Au vu de ces éléments cliniques et paracliniques, le diagnostic d’holoprosencéphalie alobaire avec diabčte insipide et hypothyroďdie centrale a été retenu. Il avait bénéficié d’un traitement quotidien ŕ base de vasopressine par voie nasale, ce qui avait entrainé une régression des signes de déshydratation et une diurčse qui était revenue ŕ la normale et d’une hormonothérapie thyroďdienne de remplacement.
L’HPE alobaire est la plus sévčre des formes classiques d’HPE. Les étiologies sont diverses regroupant les anomalies chromosomiques (trisomie 13, 18), les syndromes polymalformatifs (syndrome de CHARGE) et les facteurs environnementaux (diabčte ou hypocholestérolémie au cours de la grossesse) [1]. Les anomalies cranio-faciales les plus fréquentes sont la microcranie, l’hypotélorisme, la fente labiale médiane, l’agénésie de la mandibule… Ces anomalies ont été retrouvées par plusieurs auteurs dans la littérature [2, 3]. D’autres anomalies de la lignée médiane peuvent ętre observées ainsi un cas de sténose oropharyngée a été rapporté [4]. Le retard du développement psychomoteur constitue un élément constant dans la présentation clinique et constitue le plus souvent le motif de consultation en l’absence de malformations visibles. Dans notre cas il n’existait pas de malformations cranio-faciales. L’HPE peut s’accompagner de troubles endocriniens divers dont le plus fréquent est le diabčte insipide. Il se manifeste le plus souvent par une hypernatrémie, une polydipsie, une polyurie et une déshydratation d’intensité variable. Notre enfant avait présenté un diabéte insipide qui avait bien répondu ŕ l’administration de vasopressine par voie nasale. L’ hypernatrémie peut avoir une autre origine en cas d’holoprosencéphalie notamment une hypernatrémie neurogéne [5]. Il s’agit le plus souvent d’une hypernatrémie chronique avec adipsie sans signes de déshydratation avec une diurčse normale. Elle ne nécessite pas un traitement par de la vasopressine. La recherche d’autres troubles endocriniens chez notre enfant a permis de déceler une hypothyroďdie centrale probablement d’origine thalamique. L’IRM cérébrale qui n’a pas pus ętre réalisée chez notre enfant reste l’examen idéal pour explorer l’hypophyse et l’hypothalamus.
L’HPE est une malformation cérébrale grave due ŕ un défaut de clivage médian du prosencéphale. Chez notre enfant le diagnostic a été révélé par un retard du développement psychomoteur en l’absence de malformations cranio-faciales. Une imagerie cérébrale doit ętre réalisée devant tout retard du développement psychomoteur. Celle-ci a permis de poser le diagnostic dans notre cas. Devant toute HPE, un bilan endocrinien complet doit ętre réalisé ŕ la recherche de troubles endocriniens nécessitant un traitement particulier.
Les auteurs ne déclarent aucun conflit d’intéręt.
Ndiogou Seck a pris en charge l’enfant et écrit l’article, Idrissa Basse a participé ŕ la prise en charge de l’enfant, Younoussa Keita a aidé ŕ la bibliographie, Djibril Boiro a aidé ŕ la bibliographie, Lamine Thiam a participé ŕ la rédaction de l’article, Aliou Abdoulaye Ndongo a aidé ŕ la bibliographie, Ibrahima Diagne a supervisé et corrigé le travail.
Tableau 1: résultats bilan biologique effectué
Figure 1: coupe sagitale TDM cérébrale holoprosencéphalie alobaire avec aspect de fer ŕ cheval
Figure 2: coupe frontale TDM cérébrale: holoprosencéphalie alobaire
- Dubourg C, Lazaro L, Blayau M, Pasquier L, Durou Mr, Odent S, David V. Etude génétique de l'holoprosencéphalie. Annales de Biologie Clinique. 2003;61(6):679-687. Google Scholar
- Mansouri Hattab N, Lahmiti S, Bouaichi A, Hiroual A. Medial cleft lip: one diagnosis masking another. Arch Pediatr. 2011 Feb;18(2):149-5. PubMed | Google Scholar
- Dia Aliou Amadou, D'Almeida Franck, Mbodji Mamadou, Ka Mamadou Mourtalla. Holoprosencéphalie alobaire dans un contexte de syndrome polymalformatif: apport de l’imagerie ŕ propos d’un cas. Pan Afr Med J. 2013;15:83. Google Scholar
- Kenji Hishikawa, Hideshi Fujinaga, Chie Nagata, Masataka Higuchi, Yushi Ito. Semilobar Holoprosencephaly with congenital oropharyngeal stenosis in a term neonate. Am J Perinatol Rep. 2015;5(2):e109-e110. PubMed | Google Scholar
- Cuisset JM, Cuvellier JC, Vallée L, et al. Holoprosencephaly with neurogenic hypernatremia. Arch Pediatr. 1999 Jan;6(1):43-5. PubMed | Google Scholar
Search
This article authors
On Pubmed
On Google Scholar
Citation [Download]
Navigate this article
Similar articles in
Key words
Tables and figures

Article metrics
Recently from the PAMJ








Authors´ services
