
Syndrome de Bean chez l’enfant: ŕ propos de deux cas
Achraf El Bakkaly, Fouad Ettayebi, Houda Oubeja, Mounir Erraji, Hicham Zerhouni
Corresponding author: Achraf El Bakkaly, Service des Urgences Chirurgicales Pédiatriques, CHU Ibn Sina / Faculté de Médecine Mohammed V, Rabat, Maroc 
Received: 06 Nov 2016 - Accepted: 24 Aug 2017 - Published: 04 Oct 2017
Domain: Medical/Public health informatics
Keywords: Angiomatose, syndrôme de Bean, chirurgie
©Achraf El Bakkaly et al. Pan African Medical Journal (ISSN: 1937-8688). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: Achraf El Bakkaly et al. Syndrome de Bean chez l’enfant: ŕ propos de deux cas. Pan African Medical Journal. 2017;28:102. [doi: 10.11604/pamj.2017.28.102.11109]
Available online at: https://www.panafrican-med-journal.com//content/article/28/102/full
Original article 

Syndrome de Bean chez l’enfant: ŕ propos de deux cas
Syndrome de Bean chez l'enfant: ŕ propos de deux cas
Bean's syndrome in children: about two cases
Achraf El Bakkaly1,&, Fouad Ettayebi1, Houda Oubeja1, Mounir Erraji1, Hicham Zerhouni1
1Service des Urgences Chirurgicales Pédiatriques, CHU Ibn Sina, Faculté de Médecine Mohammed V, Rabat, Maroc
&Auteur correspondant
Achraf El Bakkaly, Service des Urgences Chirurgicales Pédiatriques, CHU Ibn Sina,
Faculté de
Médecine Mohammed V, Rabat, Maroc
L'angiomatose diffuse ou syndrome de Bean est une entité rare caractérisée par des malformations veineuses essentiellement cutanées et digestives pouvant se compliquer d'hémorragie de gravité variable. Notre travail intéresse l'étude de deux enfants, l'un âgé de 5 ans et l'autre de 9 ans et demi atteints d'angiomatose diffuse suivis au service des urgences chirurgicales pédiatriques depuis des années. Le diagnostic a été évoqué devant des rectorragies et/ou des mélénas occasionnant une anémie sévčre nécessitant des transfusions réguličres chez nos deux patients ainsi que l'apparition des angiomes cutanés au niveau des membres. Les explorations radiologiques ont révélé la présence au niveau du jéjunum et l'iléon des multiples lésions correspondant ŕ une angiomatose gręlique diffuse pour l'enfant de 9 ans et demi; elles n'ont pas retrouvé de localisations abdominales pour l'enfant de 5ans. Les deux malades ont été admis au bloc opératoire avec individualisation ŕ l'exploration des angiomes dont certains saignaient activement. Une résection par entérotomie a été réalisée. Les suites ont été marquées par l'arręt des saignements. L'intéręt de notre travail est de mettre le point sur cette entité pathologique rare, ainsi que le bénéfice du traitement chirurgical pour le contrôle des complications de cette pathologie et pour la diminution de la fréquence des transfusions.
English abstract
Diffuse angiomatosis or Bean's syndrome is a rare disease characterized by venous malformations mainly involving the skin and the digestive tract which can result in hemorrhage of variable severity. This study reports the case of two children aged 5 and 9 and a half years respectively with diffuse angiomatosis who had been treated in the Department of Emergency Paediatric Surgery over the years. The diagnosis was based on rectal bleeding and/or melenas causing severe anemia requiring regular transfusions in both patients as well as skin angiomas occurrence at the level of the limbs. Radiological evaluation showed the presence of multiple lesions at the level of the jejunum and ileum consistent with small intestinal angiomatosis in the child aged 9 and a half years. It didn't show abdominal lesions in the child aged 5 years. The two patients were admitted to the operating block. Angiomas were surgically individualized. Some of them were actively bleeding. Enterotomy was performed. Postoperative sequelae was marked by the stop of the bleedings. This study aims to update the current understanding of this rare pathology as well as the benefit of surgical treatment in controlling the complications caused by this pathology and in reducing the frequency of transfusions.
Key words: Angiomatosis, Bean's syndrome, surgery

Le syndrome de Bean est une maladie rare caractérisée par des malformations veineuses essentiellement cutanées et digestives, dont les complications hémorragiques peuvent engager le pronostic vital [1]. Ces malformations veineuses sont multifocales et touchent le plus souvent la peau et le tractus digestif, mais peuvent également ętre localisées au niveau du cerveau, des reins, des poumons, des yeux, des os et męme d'autres organes. Les lésions sont généralement présentes dčs l'enfance mais peuvent également se développer plus tard dans la vie [2]. De diagnostic difficile, nous rapportons ŕ travers ce travail l'observation de deux enfants suivis pour syndrome de Bean, entité vasculaire rare ŕ ne pas méconnaître, dont le traitement chirurgical a permis de contrôler les lésions intestinales responsables d'hémorragies et d'améliorer ainsi la qualité de vie de nos patients.

Observation n°1
Il s'agit de S.M. enfant âgé de 5 ans, deuxičme d'une fratrie de deux, bien vacciné selon le programme national de vaccination, ayant comme antécédents une communication interventriculaire trabéculaire, traitée par cerclage de l'artčre pulmonaire ŕ l'âge de 7 mois. Nous signalons une notion d'hospitalisation ŕ trois reprises au service pour prise en charge de syndrome anémique non étiqueté jusque-lŕ. Le début de la symptomatologie remonte ŕ l'âge de 3ans et 8 mois, par un saignement aprčs son cerclage de l'artčre pulmonaire, ayant nécessité une premičre transfusion. Par la suite, l'enfant a développé un syndrome anémique pour lequel il a été mis sous traitement martial, en plusieurs cures sans résultats satisfaisants, d'oů son orientation vers l'hôpital d'enfants de Rabat pour bilan étiologique. Lors de sa premičre hospitalisation, l'enfant se présentait pour un syndrome anémique franc, avec pâleur cutanéomuqueuse et asthénie avec des chiffres de numération objectivant une anémie hypochrome microcytaire hyposidéremique, pour lesquels il a bénéficié d'une transfusion de culots globulaires. Son examen clinique trouvait un enfant pâle asthénique avec ŕ l'examen cardiaque un souffle en jet de vapeur.
Lors de sa deuxičme hospitalisation pour le męme tableau clinique, pour lequel il a bénéficié d'une nouvelle transfusion, le bilan étiologique réalisé dans le cadre de la recherche d'un foyer de saignement occulte s'est avéré jusque-lŕ non concluant. Par ailleurs, l'examen clinique de l'enfant a objectivé une douleur au niveau du coude et de la plante du pied droits. L'exploration échographique couplée au doppler des deux foyers a objective la présence d'angiomes au niveau des deux sičges sans autres anomalies.Citons que par ailleurs, l'examen clinique ne rapporte pas la présence d'angiomes cutanés. Un bilan d'extension ŕ la recherche d'autres localisations, soit une imagerie par résonnance magnétique cérébrale, a objectivé la présence d'une malformation vasculaire temporale droite. L'échographie abdominale ne retrouve pas de localisations intra abdominales. Le reste des explorations étiologiques parallčles revenaient tjrs normales. L'enfant revient 20 jours aprčs dans un tableau d'anémie sévčre avec méléna nécessitant une transfusion en urgence, compliquée par l'apparition de rectorragies d'importante abondance le jour męme, avec ŕ l'examen un abdomen souple non douloureux. Un doppler abdominal réalisé en urgence parle d'une invagination gręlo-gręlique, secondaire a une masse, probablement un angiome vu le contexte clinique. Uneéchographie cardiaque réalisée aussi vu le contexte particulier de l'enfant a objectivé un état stable permettant la prise en charge chirurgicale. L'enfant est transféré par la suite au service des urgences chirurgicales pour une exploration laparoscopique convertie en laparotomie aprčs la découverte de plusieurs angiomes gręliques (Figure 1). Une résection des angiomes a été réalisée. Nous notons par ailleurs l'absence d'invagination, de diverticule du Meckel ou de masse intra abdominale. Les suites post opératoires étaient sans incidents, avec la reprise d'un bon état général et la disparition des rectorragies. L'étude anatomopathologique de la pičce opératoire parle d'un hémangiome gręlique. Avec un recul de 3 ans, l'enfant n'a plus présenté d'épisodes de saignement. Toujours sous traitement martial, il garde toujours des chiffres d'hémoglobine de contrôle corrects.
Observation n°2
Il s'agit de l'enfant O.J âgé de 9 ans et demi, l'aîné d'une fratrie de deux, bien vacciné selon le programme national d'immunisation (PNI), ayant comme antécédents une notion d'hospitalisations ŕ 4 reprises au service pour un syndrome anémique non étiqueté jusque-lŕ. Le début de la symptomatologie remonte ŕ l'âge de 4 ans oů l'enfant a présenté des rectorragies nécessitant une transfusion, il a été mis sous traitement martial sans réel rétablissement. Lors de ses hospitalisations, l'enfant a présenté un syndrôme anémiquefranc, avec un tableau clinique fait d'une pâleur cutanéo-muqueuse, asthénie et un souffle systolique ŕ l'auscultation cardiaque. Une numération formule sanguine (NFS) a été réalisée objectivant une anémie profonde hypochrome microcytaire avec des réserves ferriprives effondrées, l'enfant a bénéficié d'une réanimationhydro-électrolytique et des transfusions de culots globulaires. Par ailleurs, ŕ l'âge de 8 ans, l'enfant a présenté des douleurs au niveau du coude et de la plante du pied droits.
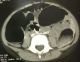
Dans le cadre d'un bilan d'extension, une tomodensitométrie et une imagerie par résonance magnétique cérébrale sont réalisées, objectivant la présence d'une malformation vasculaire cérébrale temporale droite. L'échographie abdominale n'a pas trouvé de localisations intra-abdominales. Le reste des explorations étiologiques parallčles sont revenues normales notamment une scintigraphie abdominale et une fibroscopie oeso-gastro-duodénale. L'enfant est revenu un mois aprčs, dans un tableau d'anémie sévčre avecdes rectorragies importantes nécessitant une réanimation avec une transfusion enurgence. L'examen clinique ŕ l'admission a retrouvé un enfant asthénique avec pâleur cutanéomuqueuse et une tachycardie ŕ 130 battements par minute. La tension artérielle était normale. L'écho-doppler abdominale a révélé la présence d'un boudin d'invagination gręlo-gręlique, secondaire ŕ une masse, probablement un angiome vu le contexte clinique. La tomodensitométrie abdominale est demandée, montrant la présence au niveau du jéjunum d'une masse d'environ 3cm hyper-vasculaire compatible avec un hémangiome et présence de deux autres lésions pouvant correspondre ŕ une angiomatose gręlique diffuse (Figure 2).
Aprčs la réalisation d'un bilan pré-anesthésique notamment une échographie cardiaque éliminant une contre-indication ŕ la chirurgie, l'enfant est transféré au service des urgences chirurgicales pédiatriques pour une exploration chirurgicale. L'exploration chirurgicale par voie coelioscopique est faite, l'examen du gręle a objectivé une angiomatose intestinale diffuse. Aprčs extériorisation du gręle, le geste chirurgical a consisté en une électrocoagulation des petites lésions et une résection limitée du gros angiome suivie de sutures transversales (Figure 3). L'intervention a duré 1h35min. Notre malade a bénéficié d'une exploration chirurgicale par voie coelioscopique pour une évaluation initiale aussi bien diagnostic que thérapeutique, cette voie d'abord offre l'élégance, l'efficacité, l'économie et le confort. L'étude anatomopathologique de la pičce opératoire parle d'une malformation veineuse de localisation gręlique. Les suites opératoires sont simples, le malade a quitté le service dans les 72 heures qui suivent. Le suivi ŕ long terme a objectivé une récidive des rectorragies qui a nécessité un autre séjour hospitalier dans notre service, une deuxičme exploration laparoscopique est faite avec une cure chirurgicale des deux autres gros angiomes. Les suites ont été marquées par l'arręt des saignements. L'enfant est suivi depuis lors réguličrement en consultation, avec un recul de 3 ans, ne présentant plus d'épisodes de saignement et il a été mis sous traitement martial. Les chiffres de l'hémoglobine de contrôle sont corrects. L'association de l'atteinte cutanée et digestive de ces malformations veineuses fait évoquer en premier le syndrôme de Bean, qui est retenue comme diagnostic définitif pour nos deux malades.
Le syndrôme « bluerubber-blebnaevus » ou angiomatose diffuse a été signalé pour la premičre fois en 1860, chez un patient qui a présenté une asphyxie suite ŕ une hémorragie aigue d'une tumeur parotidienne. Il avait aussi des angiomes multiples au niveau cutané etdigestif. Appelé aussi syndrôme de Bean, c'est cet auteur qui en 1958, décrit les lésions cutanées compressibles ŕ la palpation en « Tétine de caoutchouc » [3]. Plus de 200 cas ont été décrits, mais la prévalence reste inconnue. Ce syndrôme de Bean est une malformation veineuse disséminée et sporadique qui pourrait ętre familiale avec une transmission autosomique dominante secondaire ŕ une mutation sur le chromosome 9p [1], mais il semble qu'il y ait eu confusion dans la littérature avec les malformations veineuses cutanéo-muqueuses sans localisation viscérale et dont la forme familiale est liée au gčne muté « TIE2 »; le syndrome de Beann'est pas lié au gčne muté « TIE2 ». Les signes cliniques principaux découlent du saignement aigu ou chronique des multiples lésions du tractus digestif qui sičgent surtout au niveau de l'intestin gręle. Les saignements aigus se présentent comme des hématémčses, des mélénas ou des rectorragies. Les lésions sont cutanées et viscérales (atteinte digestive gastro-intestinale responsable d'hémorragies [4] et atteintes urinaire, hépatique, splénique, cérébrale, pulmonaire, etc.). Les atteintes extra-digestives peuvent ętre responsables d'une déformation orthopédique [5], une démence [6], une embolie pulmonaire, une hypertension de l'artčre pulmonaire, un hémothorax, un hémopéricarde [7], une exophtalmie [8], une thrombopénie, une coagulopathie de consommation chronique. Les lésions cutanées peuvent ętre maculaires, papuleuses, nodulaires oupédonculés, rougeâtres ou violacées. Elles sont multiples et le plus souvent nonsaignantes [9,10]. On peut les classer en 3 types [11]: 1) Le type 1: il s'agit d'une grande lésion déformante qui peut obstruer l'organe adjacent. 2) Le type 2: c'est le type le plus fréquent, il est de couleur bleue froissée en« tétine de caoutchouc » recouvert de peau [12]. 3) Le type 3: c'est une lésion maculaire ou papulaire de couleur bleu-noirequi peut blanchir ŕ la pression. Ces lésions cutanées peuvent ętre présentes ŕ la naissance ou apparaître au fil des années [9-11].
Les angiomes cutanés peuvent devenir douloureux, cette douleur est le fait de la mise en tension de ces lésions dans certaines positions, mais aussi de thromboses localisées, ŕ l'origine des phlébolithes qui sont des calcifications rondes pathognomoniques des malformations veineuses et bien visibles sur les radiographies [9,10]. L'hémorragie digestive est la complication la plus fréquente de l'atteinte intestinale dans le syndrome de Bean, mais une invagination intestinale est aussi décrite dans quelques publications [13] comme c'est le cas pour nos deux patients. Les méthodes diagnostiques comprennent la numération sanguine, l'endoscopie, l'échographie, le scanner, l'imagerie par résonance magnétique et l'histologie. Le syndrome de Bean est une forme clinique rare de malformations veineuses dont le traitement est ŕ la fois médical et chirurgical. Le pronostic de syndrome de Bean dépend des complications essentiellement hémorragiques et de la localisation de ces angiomes par rapport aux organes vitaux. En l'absence d'un traitement approprié, les patients atteints souffrent d'une anémie due aux saignements chroniques qui nécessite l'administration de fer et de transfusions ŕ répétition. La transformation maligne des lésions n'a, jusqu'ŕ présent, pas été décrite. En l'absence de saignement intestinal important, un traitement minimum est suffisant (supplémentation en fer et/ou transfusions). L'administration au long cours d'octréotide réduit les pertes sanguines du syndrôme de Bean et d'autres lésions vasculaires gastro-intestinales. Dans notre expérience, la chirurgie avec exérčse des angiomes intestinaux par voie laparoscopique ou classique, semble ętre le moyen efficace pour améliorer le pronostic des enfants atteints de ce syndrôme et réduire ainsi la fréquence des transfusions, c'est le cas pour notre deuxičme patient dont on a pu contrôler l'hémorragie causée par les angiomes intestinaux, grâce ŕ deux interventions chirurgicales, lui évitant depuis lors des transfusions répétitives. Nos résultats corroborent parfaitement avec celles de Zhi Li [14] qui admet que l'éradication chirurgicale des angiomes intestinaux est le moyen efficace pour traiter l'angiomatose de Bean. Ainsi, sur une étude faite sur 2 malades, l'auteur démontre que la chirurgie apporte le bénéfice de contrôler des saignements graves chroniques sur des angiomes intestinaux quel que soit leur nombre ou leur emplacement et ceci par résection de toutes les lésions responsables de saignement ainsi que des segments digestifs sičges de lésions de viabilité douteuse. Ce traitement couplé ŕ une supplémentation en fer seulement, a permis d'améliorer les chiffres d'hémoglobine chez les 2 patients sans ętre obligé ŕ leur refaire des transfusions. Seulement un patient a été réadmis au bloc avec exérčse des angiomes au niveau du duodénum et du côlon avec une nette amélioration clinique et des chiffres d'hémoglobine post-opératoires.
Aucun consensus n'a été établi sur le traitement de cette angiomatose diffuse. Puisque le saignement chronique des angiomes au niveau gastro-intestinal a persisté durant toute la vie des patients atteints de cette entité pathologique. Un grand choix de stratégies thérapeutiques pharmacologiques a été proposé, notamment l'interféron antiangiogenic. Cependant, ces agents n'ont pas été démontrés pour ętre fiables et efficaces pour arręter le saignement et contrôler ainsi ces angiomes gastro-intestinaux [15,16]. Récemment, Fishman et d'autres chercheurs ont conclu que les lésions de syndrôme de Bean sont des malformations vasculaires congénitales plutôt que des tumeurs de prolifération. En se basant sur cette théorie, Fishman a soigné 10 patients de syndrôme de Beanpar la chirurgie, une moyenne de 137 lésions ont été réséqués pour l'ensemble des patients, avec un peu de répétition des saignements sans anémie grave chez un seul patient, et d'avantage d'intervention chirurgicale chez deux patients [17]. Ces deux cas repris chirurgicalement ont également démontré que l'éradication chirurgicale pour les lésions gastro-intestinales de cette angiomatose était importante et efficace pour arręter définitivement le saignement de ces angiomes gastro-intestinaux. Dans notre expérience, nous croyons que le saignement chronique grave des angiomes digestifs de l'angiomatose diffuse peut ętre jugulé avec succčs par la résection de ces angiomes, indépendamment de leur nombre ou de leur emplacement. Il est peu susceptible de les traiter par les autres thérapeutiques pharmacologiques. Cette approche chirurgicale effective peut aider ŕ éviter les ravages de l'anémie chronique, y compris la fatigue, la surcharge de fer, et les risques potentiels des transfusions sanguines répétitives comprenant le risque de contamination par hépatite et accidents allergiques. L'intervention chirurgicale est la plus bien projetée quand une indication de transfusion devient évidente. Une fois une approche effective est choisie, une visualisation complčte du tractus gastro-intestinal entier de la bouche ŕ l'anus doit ętre exécutée. L'éradication complčte peut éviter le besoin de reprises chirurgicales. La résection inachevée de n'importe quelles lésions données peut tenir compte du risque de récidive et du saignement récurrent. Bien qu'extręmement pénibles, les principes standards et les techniques chirurgicaux combinés avec la gestion anesthésique soigneuse permettent un trčs bon résultat avec une morbidité minimale. Les patients avec un grand nombre de lésions peuvent avoir besoin des procédés chirurgicaux étagés. Néanmoins, cette approche améliore spectaculairement la santé et le bien-ętre des patients présentant le syndrôme de Bean. Toutefois, l'entéroscopie per-opératoire est actuellement la procédure diagnostique et/ou thérapeutique définitive puisqu'elle permet le traitement immédiat des lésions par coagulation endoscopique ou par excision chirurgicale complčte. L'indication pour cette intervention peut ętre posée ŕ partir des résultats de l'endoscopie par capsule qui permet également de contrôler ensuite les résultats du traitement [18]. Cette procédure bien qu'extręmement bénéfique, est
L'angiomatose diffuse est une malformation vasculaire rare [19]. Elle est difficile ŕ diagnostiquer en raison de sa faible fréquence. Le caractčre disséminé des lésions rend un traitement radical souvent difficile au cours du syndrome de Bean [2]. Le pronostic aprčs exérčse des angiomes intestinaux est trčs bon avec réduction de fréquence des transfusions. Cependant, le traitement endoscopique s'est montré comme une modalité thérapeutique efficace ŕ côté de la chirurgie.
Les auteurs ne déclarent aucun conflit d'intéręts.
Tous les auteurs ont contribué ŕ la rédaction de ce manuscrit, tous ont lu et approuvé la version finale.
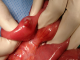
Figure 1: image en per opératoire montrant un angiome saignant activement au niveau de l'iléon
Figure 2: TDM abdominale de notre patient montrant la présence au niveau du jéjunum d'une masse d'environ 3 cm hyper-vasculaire compatible avec un hémangiome
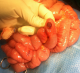
Figure 3: image en per
opératoire du gros angiome au niveau gręlique saignant (entérotomie)
- Alqahtani A, Nguyen LT, Flageole H, Laberge JM. 25 years' experience with lymphangiomas in children. J Pediatr Surg.1999;34(7):1164-1168. PubMed | Google Scholar
- Djama ML, Médaoud S, Hakem D, Berrah A. Syndrome de BEAN: ŕ propos d'une observation. La Revue de Médecine Interne. 2015;36(1):102-103. Google Scholar
- North PE, Waner M, Mizeracki A, Mrak RE, Nicholas R, Kincannon J et al. A unique microvascular phenotype shared by juvenile hemangiomas and human placenta. Arch Dermatol. 2001;137(5):559-570. PubMed | Google Scholar
- Buchi KN.Vascular malformations of the gastrointestinal tract. Surg Clin North Am. 1992;72(3):559-570. PubMed | Google Scholar
- Aihara M, Konuma Y, Okawa K, Komai R, Kudo I, Morioka R et al. Blue rubber bleb nevus syndrome with disseminated intravascular. Tohoku J Exp Med. 1991;163(2):111-117. PubMed | Google Scholar
- Wittle JT. Band ligation for colonic bleeding: modification of multiband ligating devices for use with a colonoscope. Gastrointest Endosc. 2000;52(6):762-765. Google Scholar
- Jin XL, Wang ZH, Xiao XB, Huang LS, Zhao XY. Blue rubber bleb nevus syndrome: a case report and literature review. World J Gastroenterol. 2014;20(45):17254-17259. PubMed | Google Scholar
- Rodrigues D, Bourroul ML, Ferrer AP, Monteiro Neto H, Gonçalves ME, Cardoso SR. Blue rubber bleb nevus syndrome. Rev Hosp Clin Fac Med Sao Paulo. 2000;55(1):29-34. PubMed | Google Scholar
- Zahedi MJ, Moghadam SD, Mirzaei SMS, Dehghani M, Shafiei S, Rasti A. Blue rubber bleb nevus syndrome as a rare cause of iron deficiency anemia: a case report and review of literature. Middle East J Dig Dis. 2013 Oct;5(4):235-239. PubMed | Google Scholar
- Kisu T, Yamaoka K, Lichida Y, Muri H, Nakama T, Hisatsugu T et al. A case of blue rubber bleb nevus syndrome with familial onset. Gastroenterol Jpn. 1986;21(3):262-266. PubMed | Google Scholar
- Busund B, Stray-Pedersen S, Iversen OH, Austad J. Blue rubber bleb nevus syndrome with manifestations in vulva. Acta Obstet Gynecol Scand. 1993;72(1):310-313. PubMed | Google Scholar
- Sancheti K, Podder I, Das A, Somodyuti Chandra, Gharami RC. Unilateral, linear blue rubber bleb nevus syndrome (Bean's syndrome): an unfamiliar presentation: first case from India. Indian Journal of Dermatology. 2015;60(6):616-619. Google Scholar
- Gascoyen GG. Case of naevus involving the parotid gland and causing death from suffocation: naevi of the viscera. Trans Pathol Soc London. 1860;11:267. Google Scholar
- Li Z, Yuan H, Yu D, Victor N, Chen Y, Feng J, Teitelbaum D, Sun X. The surgery for blue rubber bleb nevus syndrome. Annals of Medicine and Surgery. 2016;5(1):73. PubMed | Google Scholar
- Kopacova M, Tacheci I, Koudelka J et al. A new approach to blue rubber bleb nevus syndrome: the role of capsule endoscopy and intra-operative enteroscopy. Pediatric Surgery Int. 2007;23(7):693-697. PubMed | Google Scholar
- Okabayashi K, Hasegawa H, Nishibori H et al. A case of laparoscopic surgery for blue rubber bleb nevus syndrome. Hepatogastroenterology. 2007;54(74):451-453. PubMed | Google Scholar
- Fishman SJ, Smithers CJ, Folkman J et al. Blue rubber bleb nevus syndrome: surgical radication of gastrointestinal bleeding. Ann Surg. 2005;241(3):523-528. PubMed | Google Scholar
- Dupont-Lucas C, Bellaďche M, Mouterde O et al. Quelles indications pour l'endoscopie du gręle par vidéocapsule en pédiatrie. Archives de Pédiatrie. 2010;17(9):1264-1272. Google Scholar
- Berlyne GM, Berlyne N. Anaemia due to blue rubber bleb naevus disease. Lancet. 1960;2:1275-1277. Google Scholar
Search
This article authors
On Pubmed
On Google Scholar
Citation [Download]
Navigate this article
Similar articles in
Key words
Tables and figures
Article metrics
PlumX Metrics
Syndrome de Bean chez l’enfant: ŕ propos de deux casRecently from the PAMJ








Authors´ services
