
Découverte fortuite d'une drépanocytose hétérozygote composite S/C
Asmâa Biaz, Marwa Neji, Yousra Ajhoun, Samira EL Machtani Idrissi, Abdellah Dami, Karim Reda, Zohra Ouzzif, Sanae Bouhsain
Corresponding author: Asmâa Biaz, Service de Biochimie-Toxicologie Hôpital Militaire D’instruction Mohammed V, Rabat, Maroc 
Received: 11 May 2017 - Accepted: 16 May 2017 - Published: 07 Jun 2017
Domain: Biochemistry
Keywords: Rétinopathie, hémoglobinopathie, syndrome drépanocytaire composite SC
©Asmâa Biaz et al. Pan African Medical Journal (ISSN: 1937-8688). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: Asmâa Biaz et al. Découverte fortuite d'une drépanocytose hétérozygote composite S/C. Pan African Medical Journal. 2017;27:93. [doi: 10.11604/pamj.2017.27.93.12724]
Available online at: https://www.panafrican-med-journal.com//content/article/27/93/full
Original article 

Découverte fortuite d'une drépanocytose hétérozygote composite S/C
Découverte fortuite d'une drépanocytose hétérozygote composite S/C
Fortuitous detection of composite heterozygous S/C sickle cell disease
Asmâa Biaz1,&, Maroua Neji1, Yousra Ajhoun2, Samira EL Machtani Idrissi1,3, Abdellah Dami1,3, Karim Reda2,3, Zohra Ouzzif1,3, Sanae Bouhsain1,3
1Service de Biochimie-Toxicologie Hôpital Militaire d’Instruction Mohammed V, Rabat, Maroc, 2Service d’Ophtalmologie Hôpital Militaire d’Instruction Mohammed V, Rabat, Maroc, 3Faculté de Médecine et de Pharmacie de Rabat, Université Mohamed V Souissi, Rabat, Maroc
&Auteur correspondant
Asmâa Biaz, Service de Biochimie-Toxicologie Hôpital Militaire D’instruction
Mohammed V, Rabat, Maroc
Le syndrome drépanocytaire composite SC représente 20% ŕ 30% des syndromes drépanocytaires majeurs. Nous rapportons le cas d'une découverte fortuite d'une drépanocytose hétérozygote composite SC dans un contexte de décollement rétinien. Il s'agit d'une patiente hospitalisée au service d'ophtalmologie pour une baisse de l'acuité visuelle détectée depuis 06 mois et rebelle au traitement. Les antécédents cliniques sont dominés par la mise en place d'une prothčse totale de la hanche (PTH) douze ans auparavant. Notre observation rappelle la grande variabilité clinique de la maladie drépanocytaire imposant un dépistage précoce des patients ŕ risque avec une surveillance clinique adaptée afin d'éviter l'évolution vers des séquelles organiques irréversibles telles que la rétinopathie drépanocytaire.
English abstract
Composite S/C sickle cell disease accounts for 20%-30% of major sickle cell syndromes. We report a case of fortuitous detection of composite heterozygous S/C sickle cell disease in the context of retinal detachment. The patient had been hospitalized in the Department of Ophthalmology for treatment-resistant decreased visual acuity detected 06 months before. The patient's clinical history was marked by total hip replacement (THR) twelve years before. Our study highlights the wide clinical variability of sickle cell disease underlying the importance of early screening and adapted clinical monitoring of patients at-risk, in order to avoid its evolution toward irreversible organic sequelae such as sickle cell retinopathy.
Key words: Retinopathy, hemoglobinopathy, composite S/C sickle cell disease

Le syndrome drépanocytaire composite SC représente 20% ŕ 30% des syndromes drépanocytaires majeurs. Il est particuličrement fréquent dans certains pays de l'Afrique de l'Ouest tels que le Ghana, le Burkina Fasso, ou le Nigéria [1]. Il constitue une entité autonome trčs différente de la drépanocytose homozygote avec une atténuation des signes cliniques et une discrétion des anomalies hématologiques ŕ l'origine d'un retard de diagnostic ŕ l'âge adulte, au stade de séquelles irréversibles [2, 3]. Nous rapportons le cas d'une découverte fortuite d'une drépanocytose hétérozygote composite SC dans un contexte de décollement rétinien.

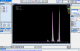
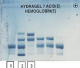
Il s'agit d'une patiente d'origine marocaine, âgée de 53 ans, mariée et mčre de 03 enfants, hospitalisée au service d'ophtalmologie pour une baisse de l'acuité visuelle détectée depuis 06 mois et rebelle au traitement. Les antécédents cliniques sont dominés par la mise en place d'une prothčse totale de la hanche (PTH) douze ans auparavant. La patiente ne dispose cependant pas d'aucun compte rendu de cette hospitalisation. L'examen de fond dœil retrouve un décollement de la rétine de l'œil droit avec des signes de vascularite rétinienne bilatérale (rétinopathie stade IV) (Figure 1). L'hémogramme objective une discrčte anémie avec un taux d'hémoglobine (Hb) ŕ 11.8 g/dL normochrome normocytaire (volume globulaire moyen (VGM) ŕ 89.2 fl et teneur corpusculaire moyenne en hémoglobine (TCMH) ŕ 30.4 pg). L'examen du frottis sanguin suspecte la présence des hématies d'aspect en faucilles. Le test de falciformation est négatif. Le dosage de l'haptoglobine et des LDH est normal. Une électrophorčse capillaire de l'hémoglobine ŕ pH alcalin est réalisée sur le systčme Capillarys 2 Flex piercing (Sebia®). Elle a révélé l'absence d'HbA, la présence d'Hb S ŕ 49.6%, Hb C ŕ 42.9%, HbA 2 ŕ 3.9% et Hb F ŕ 3.6% (Figure 2). L'électrophorčse de l'hémoglobine ŕ pH acide sur gel d'agarose sur l'automate Hydrasys 2 Scan (Sebia®) confirme la double hétérozygotie SC (Figure 3). L'interrogatoire n'a pas retrouvé de notion de co-sanguinité des parents mais l'enquęte familiale n'a pu ętre réalisée de męme que l'étude génotypique. Le diagnostic retenu est un décollement rétinien compliquant une rétinopathie chez une patiente drépanocytaire hétérozygote composite SC. La prise en charge comprend un traitement par photocoagulation et une consultation spécialisée au service d'Hématologie Clinique en vue d'une prise en charge pluridisciplinaire.
Au Maroc, toute l'épidémiologie des hémoglobinopathies reste inconnue. L'OMS estime le taux des porteurs au Maroc ŕ 6.5%; ce qui laisserait supposer l'existence de 30.000 cas de formes majeures de Thalassémie et Drépanocytose [4]. Au sein de notre institution, sur une période d'une année (résultats non publiés), sur les 246 études phénotypiques de l'hémoglobine réalisées, douze cas de drépanocytose ont été identifiés soit 4.87% dont deux soit 0.81% de drépanocytose hétérozygote composite SC. La drépanocytose hétérozygote composite SC est une entité autonome, trčs différente de la drépanocytose homozygote avec une maladie systémique moins sévčre et moins invalidante. L'atténuation des symptômes cliniques et la discrétion des anomalies hématologiques font retarder le diagnostic ŕ l'âge adulte. Chez notre patiente, seule une discrčte anémie normochrome normocytaire a été retrouvée. L'évolution de la drépanocytose hétérozygote composite SC est cependant marquée par la survenue de séquelles irréversibles dont la physiopathologie fait intervenir essentiellement l'hyperviscosité. L'ostéo-nécrose aseptique de la hanche et la rétinopathie en sont les complications les plus fréquentes [5]. Une étude réalisée en Jamaďque portant sur 166 naissances dépistées drépanocytaires hétérozygotes SC et suivis ŕ l'âge adulte a montré qu'une rétinopathie était diagnostiquée chez 43% d'entre eux [6]. Une autre étude portant sur le suivi de 106 patients SC, ayant un âge médian de 50 ans, a retrouvé comme principales complications les crises vaso-occlusives (65%), la rétinopathie (35%), l'ostéonécrose aseptique de hanche (23%) et les infarctus spléniques (19%) [7]. L'atteinte rétinienne serait en rapport avec les phénomčnes vasoocclusifs entraînant l'apparition de zones non perfusées puis les stigmates de la rétinopathie proliférative apparaissent sous la forme de néovaisseaux fragiles qui se développent ŕ la limite des zones normales et non perfusées. Les mécanismes exacts de cette vasculopathie sont mal connus mais certains facteurs de croissance vasculaires, comme le VEGF, ont été incriminés [8]. Chez notre patiente, ni la mise en place d'une PTH probablement secondaire ŕ une ostéonécrose aseptique, ni les grossesses ŕ risque n'ont permis de pousser les investigations pour poser le diagnostic du syndrome drépanocytaire composite, probablement en raison de la double discrétion des symptômes cliniques et des anomalies hématologiques. Une prise en charge précoce aurait cependant pu améliorer le pronostic fonctionnel et éviter la survenue du décollement rétinien.
Notre observation rappelle la grande variabilité clinique de la maladie drépanocytaire imposant un dépistage précoce des patients ŕ risque avec une surveillance clinique adaptée afin d'éviter l'évolution vers des séquelles organiques irréversibles telles que la rétinopathie drépanocytaire.
Les auteurs ne déclarent aucun conflit d'intéręts.
Tous les auteurs ont contribué ŕ la conduite de ce travail. Tous les auteurs déclarent également avoir lu et approuvé la version finale du manuscrit.
Figure 1: fond d’śil
Figure 2: profil électrophorétique de l’hémoglobine ŕ pH alcalin
Figure 3: electrophorčse de l’hémoglobine ŕ pH acide: (1) contrôle normal; (2) contrôle pathologique AFSC; (3) patient
- Nagel RL, Fabry ME, Steinberg MH. The paradox of hemoglobin SC disease. Blood Rev. 2003; 17(3): 167-78. PubMed | Google Scholar
- Markham MJ, Lottenberg R, Zumberg M. Role of phlebotomy in the management of hemoglobin SC disease: case report and review of the literature. Am J Hematol. 2003; 73(2): 121-5. PubMed | Google Scholar
- Bouchaďr N, Manigne P, Kanfer A, Raphalen P, de Montalembert M, Hagege I, Verschuur A, Maier-Redelsperger M, Girot R. Prevention of sickle cell crises with multiple phlebotomies. Arch Pediatr. 2000; 7(3): 249-55. PubMed | Google Scholar
- OMS. Conseil exécutif EB118/5.Thalassémie et autres hémoglobinopathies. 11 mai 2006. Google Scholar
- Condon PI, Serjeant GR. Behaviour of untreated proliferative sickle retinopathy. Br J Ophthalmol. 1980; 64(6): 404-11. PubMed | Google Scholar
- Downes SM, Hambleton IR, Chuang EL, Lois N, Serjeant GR, Bird AC. Incidence and natural history of proliferative sickle cell retinopathy: observations from a cohort study.Ophthalmology. 2005; 112(11): 1869-75. PubMed | Google Scholar
- Koduri PR, Agbemadzo B, Nathan S. Hemoglobin S-C disease revisited: clinical study of 106 adults. Am J Hematol. 2001; 68(4): 298-300. PubMed | Google Scholar
- Cao J, Mathews MK, McLeod DS, Merges C, Hjelmeland LM, Lutty GA. Angiogenic factors in human proliferative sickle cell retinopathy. Br J Ophthalmol. 1999; 83(7): 838-46. PubMed | Google Scholar
Search
This article authors
On Pubmed
On Google Scholar
Citation [Download]
Navigate this article
Similar articles in
Key words
Tables and figures

Article metrics
Recently from the PAMJ








Authors´ services
