
Un purpura thrombopénique amégacaryocytaire acquis qui cache une leucémie aigue myéloblastique
Hicham Eddou, Ali Zinebi, Abdelaziz Khalloufi, Mohammed Sina, Mehdi Mahtat, Kamal Doghmi, Mohammed Mikdame, Mohammed Karim Moudden, Mohammed El Baaj
Corresponding author: Hicham Eddou, Service de Médecine Interne, Hôpital Militaire Moulay Ismail, Meknčs, Maroc 
Received: 27 Feb 2016 - Accepted: 09 Jan 2017 - Published: 23 Jan 2017
Domain: Clinical medicine
Keywords: Amégacaryocytose acquise, purpura thrombopénique, leucémie aigüe
©Hicham Eddou et al. Pan African Medical Journal (ISSN: 1937-8688). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: Hicham Eddou et al. Un purpura thrombopénique amégacaryocytaire acquis qui cache une leucémie aigue myéloblastique. Pan African Medical Journal. 2017;26:32. [doi: 10.11604/pamj.2017.26.32.9215]
Available online at: https://www.panafrican-med-journal.com//content/article/26/32/full
Original article 

Un purpura thrombopénique amégacaryocytaire acquis qui cache une leucémie aigue myéloblastique
Un purpura thrombopénique amégacaryocytaire acquis qui cache une leucémie aigue myéloblastique
Acquired amegacaryocytic thrombocytopenic purpura hiding acute myeloid leukemia
Hicham Eddou1,2,&, Ali Zinebi1, Abdelaziz Khalloufi3, Mohammed Sina4, Mehdi Mahtat5, Kamal Doghmi5, Mohammed Mikdame5, Mohammed Karim Moudden1, Mohammed El Baaj1
1Service de Médecine Interne, Hôpital Militaire Moulay Ismail, Meknčs, Maroc, 2Faculté de Médecine et de Pharmacie de Fčs, Maroc, 3Service d’Hématologie Biologique, Hôpital Militaire Moulay Ismail, Meknčs, Maroc, 4Service d’Anatomopathologie, Hôpital Militaire Moulay Ismail, Meknčs, Maroc, 5Service d’Hématologie Clinique, Hôpital Militaire d’instruction Mohammed V, Rabat, Maroc
&Auteur correspondant
Hicham Eddou, Service de Médecine Interne, Hôpital Militaire Moulay Ismail, Meknčs, Maroc
Le purpura thrombopénique amégacaryocytaire acquis est une pathologie trčs rare caractérisé par une thrombopénie sévčre liée une réduction ou une disparition des mégacaryocytes au niveau de la moelle osseuse. Il peut ętre primaire idiopathique ou secondaire ŕ de nombreux états pathologique dont des hémopathies. Nous rapportons le cas d'un patient de 24 ans admis pour prise en charge d'un syndrome hémorragique mis sur le compte d'un purpura thrombopénique immunologique. Le diagnostic a été redressé en une amégacaryocytose aquise aprčs un échec de la corticothérapie et la réalisation d'un myélogramme. Le patient a été mis sous traitement par ciclosporine avec une évolution rapide vers une leucémie aigue myéloblastique. La progression d'une amégacaryocytose acquise vers une leucémie aigue est rapporté mais généralement pas aussi rapidement et surtout précéder par un syndrome myélodysplasique ou une aplasie médullaire. Cette observation impose un suivi strict et rapproché de ces pathologies d'apparence bénigne.
English abstract
Acquired amegakaryocytic thrombocytopenic purpura is a very rare condition characterized by severe thrombocytopenia linked to the reduction or disappearance of megakaryocytes in the bone marrow. It may be primary idiopathic or secondary to many pathological conditions including hematologic disorders. We report the case of a 24-year-old patient admitted for haemorrhagic syndrome caused by immunological thrombocytopenic purpura. The diagnosis was acquired amegakaryocytosis after the failure of corticotherapy and the performance of myelography. The patient was treated with ciclosporin with rapid progression to acute myeloblastic leukemia. The progression of acquired amegakaryocytosis to acute leukemia is reported but it is generally not so rapid and above all it is preceded by myelodysplastic syndrome or medullary aplasia. This study highlights the importance of a close follow-up of these pathologies with a benign-like appearance.
Key words: Acquired amegakaryocytosis, thrombocytopenic purpura, acute leukemia

Le purpura thrombopénique amégacaryocytaire acquis (PTAA) ou amégacaryocytose acquise, initialement décrit par Korn [1], est une entité trčs rare caractérisé par une thrombopénie sévčre liée une réduction ou une disparition des mégacaryocytes au niveau de la moelle osseuse alors que les autres lignées sont normales [2]. Cette affection peut ętre primaire ou secondaire ŕ de nombreux états pathologiques [3]. Nous rapportant dans ce travail l'observation d'un patient présentant une PTAA rapidement évoluant vers une leucémie aigue.

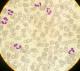
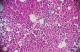
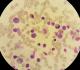
Il s'agit d'un patient de 24 ans, sans antécédents pathologiques notables, qui est admis pour prise en charge d'une épistaxis récidivante et des lésions cutanées purpuriques évoluant depuis une quinzaine de jours avec conservation de l'état général. L'examen clinique confirme la présence d'un syndrome hémorragique cutanéo-muqueux isolé. Il n'y avait pas de syndrome anémique, tumoral ou infectieux associé. L'hémogramme note une thrombopénie profonde et isolée ŕ 9 Giga/l avec des taux d'hémoglobine et de globules blancs normaux. Le frottis sanguin confirme la réduction du taux des plaquettes (Figure 1) sans anomalie morphologique des autres lignées. Le bilan d'hémostase, le bilan hépatique, le dosage des vitamines B9 et B12, les sérologies virales (Hépatite (B, C) et HIV) ainsi que le bilan immunologique (anticorps anti-nucléaire, anti DNA natif et anti-Sm) sont revenu normaux. Le diagnostic d'un purpura thrombopénique immunologique a été retenu et le patient est mis sous traitement corticoďde (bolus de méthyl-prednisolone 15mg/kg/j pendant 3 jours puis relais par prednisone ŕ la dose de 2 mg/kg/j) mais sans réponse. Cette cortico-résistance a motivé la réalisation d'un myélogramme qui a montré une moelle de richesse normal avec réduction extręme des mégacaryocytes alors que les lignées érythroblastique et granuleuses sont quantitativement et qualitativement normales (Figure 2). Une biopsie ostéo-médullaire avec étude histochimique a confirmé la diminution de la lignée mégacaryocytaire sans infiltration tumoral (Figure 3). Le scanner thoraco-abdomino-pelvien était normal. Le diagnostic a été redressé en purpura thrombopénique amégacaryocytaire acquis pour le quel le patient est mis sous traitement immunosuppresseur ŕ base de ciclosporine ŕ la dose de 5mg/kg/j avec un début de réponse dčs la 6čme semaine (taux de plaquette ŕ 50 giga/l). A 3 mois du début de traitement, le patient présente un tableau infectieux fait d'une fičvre et des frissons avec un point d'appel pulmonaire. La radiographie pulmonaire révčle un foyer basal droit. L'hémogramme montre une pancytopénie (anémie normochrome normocytaire arégénérative ŕ 90 g/l, thrombopénie ŕ 20 Giga/l et une neutropénie ŕ 0,9 Giga/l sans hyperleucocytose associée). Un frottis sanguin et médullaire montre une infiltration par 30% de blastes de taille petite ŕ moyen qui ont un rapport nucléo-cytoplsmique élevé et dont le cytoplasme est basophile renfermant quelques granulations et parfois un ou deux corps d'Auer évoquant une transformation en une leucémie aigue myéloblastique (Figure 4). La cytométrie au flux confirme le diagnostic en montrant l'infiltration par des blastes exprimant le Myéloperoxydase en intracytoplasmique, le CD117, le CD33 et le CD13. Le caryotype médullaire était normal. Le patient a bénéficié d'une chimiothérapie d'induction ŕ base de Daunorubicine et d'Aracytine compliqué d'une septicémie ŕ acinotobacter Baumani résistant ŕ une large antibiothérapie. Le décčs est survenu ŕ J18 du début de la chimiothérapie dans un tableau de choc septique.
Point du vue du biologiste: le PTAA est une maladie trčs rare caractérisée par une thrombopénie isolée qui est souvent sévčre (numération plaquettaire <20-30 × 109/L), suite ŕ une diminution marquée ou ŕ l´absence totale de mégacaryocytes dans la moelle osseuse. En en raison de sa rareté et de sa nature hétérogčne, les mécanismes pathogéniques de cette maladie ne sont pas bien définis et son étiologie est susceptible d´ętre variée. L'étude de survie des plaquettes marquées par le chrome chez des patients atteints de PTAA ne montrent aucun signe de destruction prématurée ou de séquestration. Les études de culture cellulaire in vitro ont identifié, par contre, un défaut intrinsčque des cellules progénitrices de la lignée mégacaryocytaire (CFU-MK). D'autre études suggčre une inhibition ou une destruction immunitaire des mégacaryocytes que sa soit par voie humorale ou cellulaire [4]. Le PTAA peut ętre primaire idiopathique ou survenir en association avec des troubles lymphoprolifératifs, des maladies auto-immunes (surtout un Lupus érythémateux disséminée), des infections, des tumeurs solides, des carence en vitamine B12 ou des excčs de consommation de drogues et d'alcool. Le PTAA peut également ętre la premičre manifestation de troubles de la moelle osseuse précédant un syndrome myélodysplasique ou une aplasie médullaire [4]. Une progression d'un PTAA vers une leucémie aigue est rapporté mais généralement pas aussi rapidement, comme le cas de notre observation, et surtout précéder par un syndrome myélodysplasique ou une aplasie médullaire. Dans ces derniers cas, le PTAA peut s'associer ŕ la présence d'anomalies cytogénétiques clonales acquises, une macrocytose ou des signes de dysérythropoďčse et sa différenciation avec un syndrome myélodysplasique unilignée peut s'avérer trčs difficile [5,6].
Point de vue du clinicien: sa prévalence est inconnue et la littérature reste limitée ŕ des rapports de cas. On note une légčre prédominance féminine et bien qu'il peut toucher n'importe quel âge, la plupart des cas rapportés survient chez des sujets d'âge moyens ou âgés [7]. Sur le plan clinique, le PTAA se présente habituellement par un syndrome hémorragique cutanéo-muqueux. La splénomégalie est généralement absente. L'hémogramme révčle généralement une thrombopénie isolée mais peut s'associer ŕ une anémie en cas de saignement important. A la différence du PTI, qui constitue le principal diagnostic différentiel, le myélogramme trouve une diminution importante ou une absence des mégacaryocytes. En raison de la diversité des mécanismes physiopathologiques, un large éventail de thérapies est utilisé chez les patients avec PTAA avec des degrés de réponses variables. De plus la rareté de cette pathologie ne permet pas pour le moment une conduite thérapeutique standardisée. La premičre étape du traitement consiste ŕ corriger un facteur de risque réversible, tel une infection ou un excčs de consommation d´alcool. Le recours ŕ un support transfusionnel plaquettaire peut ętre indiqué dans certaines situations hémorragiques ŕ risques notamment cérébrales. Contrairement au PTI, et comme c'est le cas de notre patient, l'utilisation d'une corticothérapie en monothérapie reste généralement inefficace pour traiter un PTAA (bien que certains groupes ont fait état de succčs avec cette approche) [8]. Les autres conduites font appel ŕ des immunosuppresseurs (cyclosporine A, cyclophosphamide, vincristine, immunoglobuline polyvalente, serum anti-lymphocytaire et le rituximab), la splénectomie ou une greffe de moelle osseuse [9]. Mais lŕ aussi la preuve d'efficacité reste faible et se base essentiellement sur des rapports de cas. L´utilisation des analogues de thrombopoietine (Romiplostin ou Eltrombopac) dans les PTAA réfractaire semble pour le moment un choix logique [10].
Le PTAA reste une pathologie rare aux mécanismes physiopathologiques intriqués et dont la conduite thérapeutique standard reste ŕ définir. L'évolution rapide, en moins de 4 mois, de notre patient vers une leucémie aigue et sans phase de myélodysplasie nous impose un suivi strict et régulier de ces pathologies d'apparence bénigne.
Les auteurs ne déclarent aucun conflit d'intéręts.
Tous les auteurs ont contribué ŕ la redaction de cet article. Les auteurs déclarent également avoir lu et confirmé la version finale de cet article.
Figure 1: frottis sanguin confirmant la présence d’une thrombopénie (coloration MGG (x100))
Figure 2: frottis médullaire montrant une cellularité normal avec absence de mégacaryocytes (coloration MGG (X 40))
Figure 3: coupe d’une biopsie ostéo-médullaire montrant une moelle de richesse normal avec absence de mégacaryocytes (HE x 100)
Figure 4: frottis médullaire montant un envahissement par des blastes granulaires (coloration MGG (x 100))
- Korn D. Congenital hypoplastic thrombocytopenia. Am J Clin Pathol. 1962 Apr;37:405-13. PubMed | Google Scholar
- Gewirtz AM, Hoffman R. Human megakaryocyte production: cell biology and clinical considerations. Hematol Oncol Clin North Am. 1990 Feb;4(1):43-64. PubMed | Google Scholar
- Trimble MS, Glynn MFX, Brain MC. Amegakaryocytic thrombocytopenia of 4 years duration: successful treatment with antithymocyte globulin. Am J Hem. 1991 ; 37 (2) : 126-127. PubMed | Google Scholar
- Lown R, Rhodes E, BosworthJ, Shannon MS, Stasi R. Acquired amegakaryocytic thrombocytopenia: potential role of thrombopoietin receptor agonists. Clin Adv Hematol Oncol. 2010 Nov;8(11):809-12. PubMed | Google Scholar
- Felderbauer P, Ritter PR, Mattern D, Schmitz F, Bulut K, Ansorge N et al. Acquired pure megakaryocytic aplasia: a separate haematological disease entity or a syndrome with multiple causes. Eur J Haematol. 2004 Jun;72(6):451-4. PubMed | Google Scholar
- Lai DW, Loughran TP, Maciejewski JP, Sasu S, Song SX, Epling-Burnette PK et al. Acquired amegakaryocytic thrombocytopenia and pure red cell aplasia associated with an occult large granular lymphocyte leukemia. Leuk Res. 2008 May;32(5):823-7. PubMed | Google Scholar
- Erkurt MA, Kaya E, Baran M, Tmen EY, Fienel S, Kuku R et al. Rapid progression of acquired amegakaryocytic thrombocytopenia to myelodysplastic syndrome: case report. Turk J Haematol. 2005;22(4): 205-208. PubMed | Google Scholar
- Mulroy E, Gleeson S, Chiruka S, Danazol. Effective option in acquired Amegakaryocytic thrombocytopaenic purpura; Case Rep. Hematol. 2015;2015:171253. PubMed | Google Scholar
- Mirzania M, Khalili S, Hasanpoor A, Shamshiri RA. Anti-CD20 antibody is effective in the patient with refractory amegakaryocytic thrombocytopenia, 25 months follow up. Int J Hematol Oncol Stem Cell Res. 2014; 8(2): 41-44. PubMed | Google Scholar
- Cela I, Miller IJ, Katz RS, Rizman A, Shammo JM .Successful treatment of amegakaryocytic thrombocytopenia with eltrombopag in a patient with Systemic Lupus Erythematosus (SLE). Clin Adv Hematol Oncol. 2010 Nov;8(11):809-12. PubMed | Google Scholar
Search
This article authors
On Pubmed
On Google Scholar
Citation [Download]
Navigate this article
Similar articles in
Key words
Tables and figures

Article metrics
Recently from the PAMJ








Authors´ services
