
Une tumeur maligne des gaines des nerfs périphériques compliquant la maladie de Von Recklinghausen
Tilila Hajjad, Samir El Mazouz, Noureddine Gharib, Abdellah El Abbassi
Corresponding author: Tilila Hajjad, Service de Chirurgie Plastique, Reconstructrice et Esthétique, CHU Ibn Sina, Rabat, Maroc 
Received: 10 May 2015 - Accepted: 18 May 2015 - Published: 03 Jul 2015
Domain: Public Health
Keywords: MPNST, neurofibromatose de type 1, chirurgie
©Tilila Hajjad et al. Pan African Medical Journal (ISSN: 1937-8688). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: Tilila Hajjad et al. Une tumeur maligne des gaines des nerfs périphériques compliquant la maladie de Von Recklinghausen. Pan African Medical Journal. 2015;21:176. [doi: 10.11604/pamj.2015.21.176.7035]
Available online at: https://www.panafrican-med-journal.com//content/article/21/176/full
Original article 

Une tumeur maligne des gaines des nerfs périphériques compliquant la maladie de Von Recklinghausen
Une tumeur maligne des gaines des nerfs périphériques compliquant la maladie de Von Recklinghausen
Tilila Hajjad1,&, Samir El Mazouz1, Noureddine Gharib1, Abdellah El Abbassi1
1Service de Chirurgie Plastique, Reconstructrice et Esthétique, CHU Ibn Sina, Rabat, Maroc
&Auteur correspondant
Tilila Hajjad, Service de Chirurgie Plastique, Reconstructrice et Esthétique, CHU Ibn Sina, Rabat, Maroc
Les tumeurs malignes des gaines des nerfs périphériques ou MPNST (Malign Peripheral Nerve Sheath Tumors selon les anglo-saxons) sont des tumeurs rares qui constituent la principale complication des neurofibromatoses de type 1 (NF1) ŕ l'âge adulte. Du fait de leur rareté ces tumeurs posent ŕ la fois des problčmes diagnostiques et thérapeutiques. Nous rapportons une nouvelle observation de MPNST survenue chez une patiente suivie pour une NF1 et dont le traitement a consisté en une exérčse large suivi d'une radiothérapie. Le but de ce travail est d'étudier les critčres diagnostiques ainsi que la prise en charge thérapeutique de ce type de tumeurs.
Les tumeurs malignes des gaines des nerfs périphériques ou MPNST (Malign Peripheral Nerve Sheath Tumors selon les anglo-saxons) constituent un groupe hétérogčne de tumeurs malignes qui dérivent des cellules qui constituent les gaines des nerfs périphériques. Ce sont des tumeurs rares et constituent la principale complication des neurofibromatoses de type 1 (NF1) ŕ l'âge adulte. Les autres MPNST sont soit radio-induites ou solitaires. Nous rapportons dans cet article une observation de MPNST compliquant une NF1.
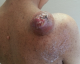
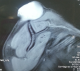
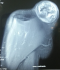
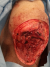
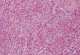
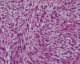
F.A, est une patiente âgée de 29 ans, suivie pour maladie de Von Recklinghausen dont la mčre et le grand-pčre sont porteurs de la męme maladie. La patiente a présenté une tuméfaction au niveau de la région scapulaire droite (Figure 1), augmentant progressivement de volume, évoluant vers l'ulcération et ce en l'espace de 3 mois. A l'examen, la patiente présente des taches café au lait sur pratiquement tout le corps, accentués au niveau du tronc avec présence de nombreux neurofibromes plexiformes au niveau du thorax et de l'abdomen. La tumeur de la région scapulaire était superficielle, mobile par rapport au plan profond, sphérique, mesurant 5cm de diamčtre avec une ulcération cutanée apicale. Une biopsie de la masse a été réalisée dont le résultat était en faveur d'une MPNST. L'IRM de l'épaule droite réalisée pour apprécier l'extension locale de la tumeur montrait la présence, au niveau des parties molles superficielles et postérieures de l'aplomb de l'épaule droite, d'une masse oblongue bien limitée de 46 x 34 mm, se développant en dehors avec respect du revętement cutané. Cette masse est de signal intermédiaire finement inhomogčne T1, discrčtement plus intense en T2 et se rehaussant de façon intense en périphérie et hétérogčne au niveau de son centre. Cette lésion refoule en dedans et en bas le muscle supra-épineux qui est intčgre. L'aspect IRM était compatible avec une MPNST (Figure 2 et Figure 3). La TDM thoraco-abdomino-pelvienne réalisée pour apprécier l'extension générale de la tumeur n'a montré aucune localisation secondaire. La patiente a subit une exérčse chirurgicale de sa tumeur avec 3 cm de marges emportant une partie du trapčze et du muscle supra-épineux ainsi que le périoste recouvrant l'épine de l'omoplate (Figure 4). L'examen histologique retrouvait une prolifération tumorale maligne constituée par une alternance de zones peu cellulaires et de zones plus cellulaires. Ces cellules sont fusiformes avec des atypies cytonucléaires modérées ŕ marquées et un index mitotique élevé. On note la présence de quelques foyers de nécrose tumorale. L'examen immunohistochimique montre que ces cellules tumorales expriment faiblement et de façon hétérogčne la PS100 (polyclonal-DAKO). L'aspect histologique et immunohistochimique sont en faveur d'une MPNST mesurant 5cm de grand axe, de grade 3 selon la classification de la FNCLCC avec des limites d'exérčse saines (Figure 5 et Figure 6). La patiente a par la suite bénéficié d'une thérapie par pression négative au niveau de la perte de substance pour favoriser le bourgeonnement suivie d'une couverture par greffe de peau semi épaisse. Elle a ensuite été adressée en radiothérapie pour complément de prise en charge.
Les tumeurs malignes des gaines des nerfs périphériques ou MPNST (Malign Peripheral Nerve Sheath Tumors selon les anglo-saxons) sont définis par la classification de l'OMS 2013 comme étant des tumeurs malignes développées [1]: soit ŕ partir d'un nerf périphérique; soit d'une tumeur bénigne des gaines des nerfs; ou chez un patient présentant une neurofibromatose de type 1 (NF1). En dehors de ces situations, le diagnostic est basé sur une association de données histologiques, phénotypiques et ultra structurales montrant une différenciation schwannienne. Il s'agit de tumeurs rares qui représentent 5 ŕ 10% des sarcomes [2]. L'incidence dans la population générale est de 0,001% et passe ŕ 5-10% chez les patients atteints de NF1 [3]. Ces tumeurs surviennent dans 50% des cas chez les patients atteints de NF1, dans 40% des cas selon un mode sporadique et seule 10% sont radio-induites [3]. Elles apparaissent généralement entre la troisičme et la quatričme décade mais une révélation plus précoce est possible particuličrement chez les patients atteints de NF1 [2]. Le sex ratio est proche de 1 avec une légčre prédominance féminine [2]. Du point de vue topographique, elles se développent essentiellement au niveau des racines des membres et du tronc (nerf sciatique, plexus brachial et plexus sacré) plus rarement au niveau de la tęte et du cou [3]. Sur le plan pathogénique, il est actuellement établi que c'est le gčne NF1 (situé sur le bras long du chromosome 17) et codant pour la protéine « neurofibromine » qui intervient dans la genčse des MPNST [4]. Cliniquement, les douleurs spontanées et le déficit neurologique sensitivomoteur font craindre la malignité. De męme que la croissance brutale d'un neurofibrome connu et stable chez un patient atteint de NF1 doit faire évoquer le diagnostic [2]. Du point de vue radiologique, les examens d'imagerie ont un double objectif: faire la distinction entre les MPNST et les tumeurs bénignes et préciser l'extension locale et générale de ces tumeurs. L'IRM est l'examen de choix pour la caractérisation des MPNST et les 4 caractéristiques qui orientent vers une MPNST plutôt que vers un neurofibrome sont [5]: une taille ≥ 5 cm; un rehaussement périphérique; des zones œdémateuses périlésionnelles; et des zones kystiques intra-tumorales (hémorragies ou nécrose) Si 2 ŕ 4 des critčres ci dessus sont présents: probabilité élevée de malignité (spécificité 90%, sensitivité 61%); pour les patients porteurs d'une NF1: une hétérogénéité en T1 est également en faveur d'une lésion maligne. Le diagnostic de certitude des MPNST est apporté par l'histologie. Macroscopiquement, elles se présentent sous forme d'une masse fusiforme englobant un nerf.
En général elles mesurent plus de 5 cm et sont de coloration blanchâtre avec des zones de nécroses et d'hémorragie. L'aspect histologique est trčs hétérogčne car les cellules présentent des degrés variables de différenciation schwannienne, fibroblastique ou périneuriale [2]. Les critčres de malignité incluent l'invasion des structures de voisinage, les emboles vasculaires, le pléomorphisme nucléaire, la nécrose et la présence de mitoses [4]. Sur le plan immunohistochimique, il n'y a pas de marqueurs spécifiques de MPNST. Cependant plusieurs marqueurs sont souvent utilisés afin de différencier ces tumeurs des autres diagnostics différentiels (mélanome, fibrosarcome, synovialosarcome monophasique, léiomyosarcome ou plus rarement neurofibrome et schwanome cellulaire)[3].Les marqueurs les plus souvent utilisés sont la protéine S100 qui est positive dans 50% des cas (mais seulement de maničre focale), la Leu-7 qui est positive dans 50% des cas, la protéine myéline basique qui est positive dans 50% des cas alors que le HMB45 et le cytokératine sont négatifs [3]. Sur le plan thérapeutique, la chirurgie reste le traitement référence des MPNST [3]. Il est primordial de réaliser une exérčse de toute la lésion avec des marges chirurgicales saines pour éviter les récidives locales. La radiothérapie néo-adjuvante ou adjuvante améliore le contrôle local et diminue le risque de récidive mais ne semble pas avoir d'effet sur la survie globale [3]. La chimiothérapie quant ŕ elle ne semble pas améliorer la survie [3]. Elle est souvent réalisée chez les patients avec des tumeurs volumineuses jugées inextirpables ou chez les patients métastatiques. L'évolution générale dépend de la taille de la tumeur, sa localisation, son grade histologique, de l'association ou non ŕ une NF1 et de la possibilité et de la qualité de la premičre résection [4]. Dans tous les cas, le pronostic est mauvais avec une survie globale ŕ 5 ans de 25% en cas de NF1 et de 50% en cas de tumeur isolée [4]. Les récidives locales sont fréquentes ainsi que les métastases (poumons, foie, peau, os) qui apparaissent dans un délai moyen de 2 ans [4].
Les MPNST sont des tumeurs rares qui représentent la principale complication des NF1 d'oů l'intéręt d'une surveillance rapprochée chez ces patients. La croissance brutale d'un neurofibrome connu et stable chez un patient atteint de NF1 doit faire évoquer le diagnostic. L'IRM est l'examen d'imagerie de choix pour faire la distinction entre les MPNST et les autres diagnostics différentiels. Le diagnostic définitif est établi par l'histologie. La chirurgie reste la pierre angulaire du traitement et la qualité de la premičre résection constitue un facteur pronostique déterminant. La radiothérapie améliore le contrôle local mais le pronostic reste péjoratif.
Les auteurs ne déclarent aucun conflit d'intéręts.
Tous les auteurs ont contribué ŕ la conduite de ce travail de la discussion du cas, l'intervention chirurgicale et la rédaction de l'article. Tous les auteurs ont contribué ŕ la conduite de ce travail. Tous les auteurs déclarent également avoir lu et approuvé la version finale du manuscrit.
Figure 1: tuméfaction au niveau de la région scapulaire droite ulcérée avec présence de taches café au lait
Figure 2: IRM de l’épaule
montre au niveau des parties molles superficielles et postérieures de l’aplomb
de l’épaule droite, une masse oblongue bien limitée de 46 x 34 mm, se développant
en dehors avec respect du revętement cutané et refoule en dedans et en bas le
muscle supra-épineux
Figure 3: aspect IRM montrant la masse prenant le contraste de façon intense en périphérie et hétérogčne au niveau de son centre
Figure 4: image postopératoire montrant la perte de substance
Figure 5: prolifération tumorale maligne d’architecture fasciculée constituée d’une alternance de zones peu cellulaires et de zones plus cellulaires (HES x100)
Figure 6: prolifération tumorale maligne faites de cellules fusiformes avec un index mitotique élevé et atypies cytonucléaires marquées (HES x 400)
- Le Guellec Sophie. Les tumeurs des gaines des nerfs périphériques. Ann Pathol. 2015 Jan;35(1):54-70. PubMed | Google Scholar
- Bouvier C, Maues de Paula A, Roche PH, Chagnaud C, Figarella-Branger D. (2013) Tumeurs du systčme nerveux périphérique. EMC-Neurologie. 2013;10:1-11. Online publication date 1-Jan-2013. Google Scholar
- Beer, Timothy C. Malignant Peripheral Nerve Sheath Tumor (MPNST): an overview with emphasis on pathology imaging and management strategies.2012. PubMed | Google Scholar
- Charfeddine I et al. Tumeur maligne des gaines nerveuses périphériques révélant une neurofibromatose type 1. Journal Tunisien d'ORL et de Chirurgie Cervico-Faciale. 2008;20:1. PubMed | Google Scholar
- Wasa, Junji et al. MRI features in the differentiation of malignant peripheral nerve sheath tumors and neurofibromas. American Journal of Roentgenology. 2010;194(6):1568-1574. PubMed | Google Scholar
Search
This article authors
On Pubmed
On Google Scholar
Citation [Download]
Navigate this article
Similar articles in
Key words
Tables and figures
Article metrics
Recently from the PAMJ








Authors´ services
