
A propos d’un rare cas de tumeur Rhabdoďde Teratoďde atypique du systeme nerveux central chez une femme enceinte
Mohammed Afif, Jihane Khalil, Fadila Kouhen, Abdellah Aissa, Youssef Omour, Mustapha Elkabous, Hanan Elkacemi, Tayeb Kebdani, Noureddine Benjaafar
Corresponding author: Mohammed Afif, Service d’Oncologie Médicale, Institut National d’Oncologie, Université Mohammed V, Rabat, Maroc 
Received: 03 Dec 2014 - Accepted: 13 Dec 2014 - Published: 05 Jan 2015
Domain: Clinical medicine
Keywords: Tumeur rhabdoide tératoide atypique, cervelet, femme enceinte
©Mohammed Afif et al. Pan African Medical Journal (ISSN: 1937-8688). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: Mohammed Afif et al. A propos d’un rare cas de tumeur Rhabdoďde Teratoďde atypique du systeme nerveux central chez une femme enceinte. Pan African Medical Journal. 2015;20:2. [doi: 10.11604/pamj.2015.20.2.5877]
Available online at: https://www.panafrican-med-journal.com//content/article/20/2/full
Original article 
A propos d’un rare cas de tumeur Rhabdoďde Teratoďde atypique du systeme nerveux central chez une femme enceinte
A propos d’un rare cas de tumeur Rhabdoďde Teratoďde atypique du systeme nerveux central chez une femme enceinte
Mohammed Afif1,&, Jihane Khalil1, Fadila Kouhen1, Abdellah Aissa1, Youssef Omour2, Mustapha Elkabous3, Hanan Elkacemi1, Tayeb Kebdani1, Noureddine Benjaafar1
1Service de Radiothérapie, Institut National d’Oncologie, Université Mohammed V, Rabat, Maroc, 2Service de Radiologie, Institut National d’Oncologie de Rabat, Université Mohammed V, Rabat, Maroc, 3Service d’Oncologie Médicale, Institut National d’Oncologie, Université Mohammed V, Rabat, Maroc
&Auteur correspondant
Mohammed Afif, Service d’Oncologie Médicale, Institut National d’Oncologie, Université Mohammed V, Rabat, Maroc
Les tumeurs rhabdoďdes tératoďdes atypiques du systčme nerveux central sont des tumeurs pédiatriques rares et de mauvais pronostic. La littérature rapporte une dizaine de cas chez l'adulte dont deux survenus au cours d'une grossesse. Nous rapportons dans ce travail, le cas d'une femme de 25 ans, enceinte de 14 semaines d'aménorrhée, qui a été opérée pour une tumeur rhabdoďde tératoďde atypique de la fosse cérébrale postérieure. Un complément thérapeutique a été discuté chez la patiente aprčs interruption thérapeutique de grossesse, mais la patiente fut décédée avant de démarrer le traitement adjuvant. Nous décrivons bričvement les caractéristiques des tumeurs rhabdoďdes, et les particularités de sa prise en charge chez l'adulte.
Les tumeurs rhabdoďdes dites également tératoďdes atypiques (TRTA) du systčme nerveux central (SNC) sont des tumeurs pédiatriques rares et agressives. Les cas rapportés chez l'adulte sont rares [1], et les cas décrits chez la femme enceinte sont exceptionnels [2, 3]. Ainsi, notre cas représente le 3čme cas décrit chez une femme enceinte.
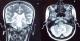
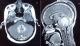
Il s'agit d'une femme de 25 ans, sans antécédents pathologiques notables, primipare et enceinte de 14 semaines d'aménorrhée , qui a présenté 3 semaines avant le diagnostic de la tumeur, un syndrome d'hypertension intracrânienne associé ŕ une baisse de l'acuité visuelle bilatérale, et un syndrome cérébelleux. Une imagerie par résonnance magnétique (IRM) cérébrale a été réalisée en urgence, et avait objectivé un processus sous tentoriel, de sičge médian, occupant le vermix, infiltrant les hémisphčres cérébelleux latéralement, et occupant le V4 avec refoulement du tronc cérébral, ce processus mesurant 56x46x40 mm, est tissulaire, hétérogčne, en hyposignal en T1, hyper signal en T2, et prend fortement le contraste en T1 aprčs injection du gadolinium (Figure 1 et Figure 2). Il est associé ŕ une hydrocéphalie, et un engagement amygdalien. La patiente a bénéficié initialement en urgence d'une dérivation ventriculopéritonéale, puis secondairement d'une exérčse large et fragmentée de la tumeur.
L'étude anatomopathologique avait objectivé un processus tumoral d'architecture polymorphe, constitué de nappes cellulaires blastémateuses, faites de cellules indifférenciées ŕ noyaux hyperchromatiques excentrés et comportant des inclusions intracytoplasmiques, ces nappes étaient séparées par un tissu mésenchymateux trčs dense comportant des cellules rhabdomyoblastiques regroupées en amas. L'aspect était en faveur d'une tumeur rhabdoďde tératoďde atypique, grade IV de l'OMS (Figure 3). L'immunohistochimie a confirmé le diagnostic avec une positivité de la vimentine; EMA, et la synaptophysine. La patiente a été adressée au service de radiothérapie pour une radiothérapie adjuvante, les options thérapeutiques ont été discutées avec la famille, et une interruption thérapeutique de grossesse ŕ été indiquée, mais la patiente fut décédée dans les suites opératoires, par détresse respiratoire, trois semaines aprčs la chirurgie, et avant de démarrer le traitement adjuvant.
Les TRTA du SNC constituent une entité pathologique rare, survenant principalement chez l'enfant, avec une fréquence ne dépassant pas 1 ŕ 2% des tumeurs cérébrales pédiatriques [4]. Les cas décrits chez l'adulte sont trčs rares, et seulement deux cas de TRTA chez une femme enceinte ont été publiés jusqu'ŕ nos jours [2, 3]. Décrites pour la 1čre fois en 1978 par Beckwith et Palmer comme une tumeur primitive rénale extręmement agressive [5], le 1er cas de tumeur rhabdoďde maligne cérébrale a été diagnostiqué chez un nourrisson âgé de trois mois par Briner (1985) [6]. La symptomatologie clinique des TRTA n'est pas spécifique, et varie en fonction de l'âge, et du sičge de la tumeur (Syndrome d'HTIC, crises convulsives, syndrome cérébelleux ou pyramidal...). La symptomatologie de l'adulte peut ętre plus accentuée, et amčne ŕ un diagnostic plus précoce [1, 7]. L'IRM est l'examen clé au diagnostic notamment chez la femme enceinte, ŕ l'IRM, les tumeurs apparaissent isointenses par rapport au cortex en T2; associées ŕ des calcifications, et des remaniements hémorragiques et kystiques, cet aspect peut poser un problčme de diagnostic différentiel avec les médulloblastomes, ce qui était le cas pour notre patiente [8]. Le diagnostic de certitude est histologique, il s'agit d'une prolifération cellulaire diffuse, faite de cellules rhabdoďdes typiques polygonales ou rondes, ŕ cytoplasme abondant, ces cellules sont sičge d'inclusions éosinophiles hyalines ŕ bordure bien définie, refoulant un noyau excentré vésiculeux, avec nucléole proéminent, et montrant des atypies marquées. Dans 85 % des cas, le contingent rhabdoďde s'associe ŕ d'autres éléments tissulaires.
L'immunohistochimie présente un grand intéręt pour la confirmation diagnostique, les TRTA expriment de la vimentine, l'EMA, et de la cytokératine, elles présentent une positivité variable pour la GFAP, la NSE, la protéine S100 et la Synaptophysine [9, 10]. Sur le plan moléculaire, les TRTA sont associées ŕ une délétion du gčne hSNF5/INI1 (gčne suppresseur de tumeur), situé au niveau du chromosome 22q11 [11, 12], cette mutation du gčne hSNF5/INI1 est pathognomonique des tumeurs avec composante rhabdoďde, et pourra ętre demandée dans les cas de difficultés diagnostiques [12]. L'exérčse chirurgicale la plus complčte semble ętre le seul traitement ayant permis des survies prolongées. La radiothérapie cérébrospinale adjuvante est recommandée ŕ cause des disséminations méningées fréquentes [1, 13]. La dose recommandée est de 36 Gy sur l'axe cérébrospinal, avec une surimpression de 24 Gy sur le lit tumoral. Certains auteurs recommandent une désescalade des doses ŕ moins de 50 Gy, afin de réduire la toxicité neurologique, sans différence sur la survie globale et la survie sans récidive. La chimiothérapie intrathécale ŕ base de cytarabine a également permis une amélioration clinique [14]. Dans les deux cas de TRTA chez une femme enceinte publiés dans la littérature, les patientes avaient une grossesse assez évoluée, l'une ŕ 33 SA, et la 2čme ŕ 36 SA, elles ont été opérées pendant la grossesse, et elles ont pu bénéficier de radiothérapie adjuvante aprčs extraction du fœtus [2, 3]. Dans notre cas, la patiente était encore au 1er trimestre de grossesse (14 SA), et une interruption thérapeutique s'avérait nécessaire avant toute décision d'un traitement adjuvant. Le pronostic des TRTA reste mauvais, avec une médiane de survie dans les séries publiées ne dépassant pas les 8 mois [1], ceci est du au caractčre agressif de la tumeur, et son potentiel métastatique élevé. Le pronostic chez l'adulte semble plus mauvais, mais la rareté des cas ne permet pas de confirmer l'impact de l'âge sur le pronostic.
L'observation que nous avons décrit confirme le caractčre agressif des tumeurs rhabdoďdes tératoďdes atypiques de l'adulte, et l'association avec la grossesse dans notre contexte a posé des difficultés pour continuer le traitement adjuvant. Une prise en charge multidisciplinaire impliquant neurochirurgien, oncologue radiothérapeute, et obstétricien est indispensable afin de mieux gérer des cas similaires.
Les auteurs ne déclarent aucun conflit d'intéręts.
Mohammed Afif ŕ fourni l'effort intellectuel pour la rédaction de l'article, Jihane Khalil, Fadila Kouhen, Abdellah Aissa, Youssef Omour, et Mustapha Elkabous ont participé ŕ la préparation des données bibliographiques et la relecture de l'article, Hanan Elkacemi, Tayeb Kebdani, et Noureddine Benjaafar ont participé ŕ la révision critique du contenu intellectuel du document et ont donné l'approbation finale de la version ŕ publier.Tous les auteurs ont contribué ŕ la conduite de ce travail. Tous les auteurs déclarent également avoir lu et approuvé la version finale du manuscrit.
Figure 1: images IRM en coupe frontale «A» et sagitale «B», objectivant une tumeur siégeant au niveau du vermix, avec un hypersignal en T2
Figure 2: images IRM T1 aprčs injection de Gadolinium (coupe axiale «A» et sagittale «B»), objectivant la tumeur du vermis avec une forte prise de contraste
Figure 3: aspect anatomopathologique de la tumeur: prolifération tumorale d’architecture polymorphe «A»; faite de cellules rhabdoďdes rondes, ŕ cytoplasme éosinophile, comportant des inclusions hyalines intracytoplasmiques «B»
- Mirone G, Bouazza S, Chibbaro S, Bresson D, Pavlika M, George B. Primary malignant rhabdoid tumour of the brain in adults. Case Reports / Journal of Clinical Neuroscience. 2009 Nov; 16 (11): 1495-1497. PubMed | Google Scholar
- Lutterbach J, Liegibel J, Kock D, Madlinger A, Frommhold H, Pagenstecher A. atypical teratoid/rhabdoid tumors in adult patients: case report and review of the literature. J Neurooncol. 2001 Mars; 52 (1): 49-56. PubMed | Google Scholar
- Erickson M, Johnson R, Bannykh S, De Lotbiniere A, Kim JH. Malignant rhabdoid tumor in a pregnant adult female: literature review of central nervous system rhabdoid tumors. Journal of Neuro-Oncology. 2005 Sep; 74 (3): 311-319. PubMed | Google Scholar
- Lee YK, Choi CG, Lee JH. Atypical teratoid/rhabdoid tumor of the cerebellum: Report of two infantile cases. ANJR Am J Neuroradiol. 2004 Mar; 25(3): 481-483. PubMed | Google Scholar
- Beckwith JB, Palmer NF. Histopathology and prognosis of Wilms tumour: results from the first national Wilms tumour study pathology center. Hum Pathol.1978 ; 14 (5) : 481-492. PubMed | Google Scholar
- Lefkowitz IB, Rorke LB, Packer RJ, Sutton LN, Segal K, Katnick RJ. Atypical teratoid tumor of infancy: definition of an entity. Ann Neurol. 1987 Jul; 22 (1): 448-449. PubMed | Google Scholar
- Arrazola A, Pedrosa I, Mendez R, Saldana C, Scheithauer BW, Martinez A. Primary malignant rhabdoid tumour of the brain in an adult. Neuroradiology. 2000 Mai; 42 (5): 363-367. PubMed | Google Scholar
- Lee IH, Yoo SY, Kim JH, Eo H, Kim OH, Cheon JE, and al. Atypical teratoid/rhabdoid tumors of the central nervous system: imaging and clinical findings in 16 children. Clinical Radiology. 2009 Mar; 64(3) : 256-264. PubMed | Google Scholar
- Judkins, AR, Eberhart, CG, Wesseling, P. Atypical teratoid/rhabdoid tumour. WHO classification of tumours of the central nervous system. 2007: 147-149. PubMed | Google Scholar
- Harmouch A, Bellarbi S, Derkaoui HF, Maher M, Elkhamlichi A, Sefiani S. La tumeur tératoďde rhabdoďde atypique cérébrale: ŕ propos de deux cas. Neurochirurgie. 2012 Aug ; 58 (4): 254-257. PubMed | Google Scholar
- Biegel JA, Tan L, Zhang F, Wainwright L, Russo P, Rorke LB. Alterations of the hSNF5/INI-1 gene in central nervous system atypical teratoid/rhabdoid tumors and renal and extrarenal rhabdoid tumors. Clin Cancer Res. 2002 Nov; 8 (11): 3461-7. PubMed | Google Scholar
- Svenet N, Sheridan E, Amram D, Schneider P, Handgretinger R, Dellatre O. Constitutional Mutations of the hSNF5/INI1 Gene Predispose to a Variety of Cancers. Am J Hum Genet. 1999 Nov; 65(5): 1342-1348. PubMed | Google Scholar
- Pimentel J, Silva R, Pimentel T. Primary malignant rhabdoďde tumors of the central nervous system: considerations about two cases of adulthood presentation. Journal of NeuroOncology. 2003; 61 (2): 121-126. PubMed | Google Scholar
- Kevin FG, Amar G. Atypical teratoid rhabdoid tumor: current therapy and future directions. Frontiers in oncology. 2012 Sep; 12 (2): 114. PubMed | Google Scholar
Search
This article authors
On Pubmed
On Google Scholar
Citation [Download]
Navigate this article
Similar articles in
Key words
Tables and figures

Article metrics
Recently from the PAMJ








Authors´ services
